L-tert-Leucine
This is a tutorial for intermediate level users. To comprehend it, you will need to know how to:
- Build a system to run molecular dynamics (MD) with Amber.
- Submit calculations using Gaussian.
This tutorial was designed as an alternative (or as a supplement) to the official Amber tutorials for parameterizing a custom residue. It summarizes information from three Amber tutorials:
I invested a considerable amount of time following Amber’s recipes to parameterize custom residues. I achieved my goal by combining parts of them to create my own protocol. I hope it also helps you. As I mentioned, this is a recipe; there is no technical or theoretical information included. That part is well explained in the Amber tutorials.
This is not the only or the best method, but it is one that works.
This tutorial can be viewed from a bird’s eye perspective by advanced users who are just looking for a few lines or commands to remember (or to tweak something in their regular workflow). Alternatively, beginners can follow along at a slower pace by reading the basics of what is being done. If additional information is needed, links to explanations will be included.
I) Residue Structure Selection
To calculate the partial atomic charges, we will need a 3D representation of our non-standard residue. For this, we can:
- Build it using, Gaussview, Maestro or any other software you are familiar with.
- Obtain the atomic coordinates from a PDB file or any other source that includes 3D information about the molecule (This option is highly recommended. It helps to maintain consistency with PDB naming schemes, and it makes your parameters easier for other people to use and understand).
In this case, the residue to be parameterized is L-tert-Leucine, which I refer to as LTL. However, the side chain can be anything you desire.
Keep in mind:
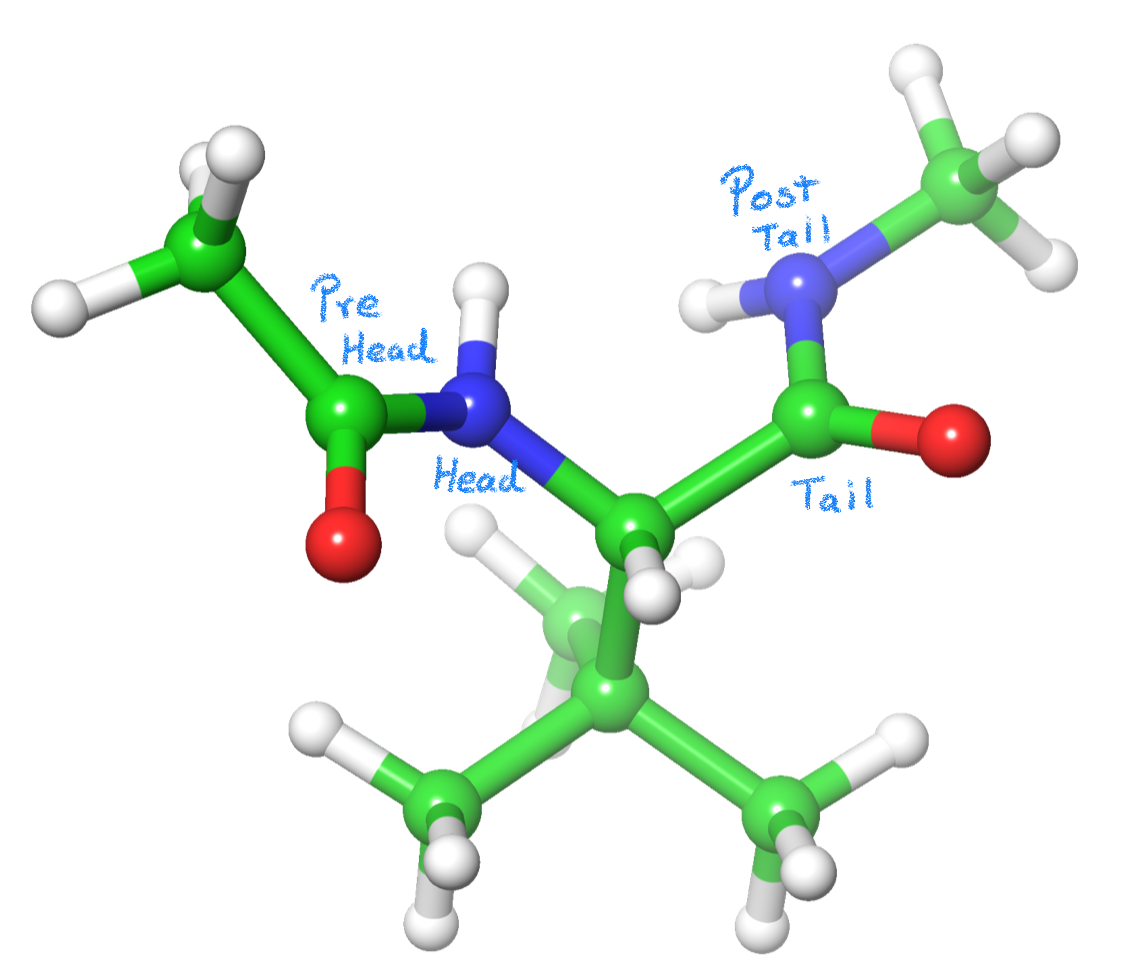
In a amino acid C=O is the tail and the NH is the head. (Fig. 1)
When naming your custom residues, use three or four capital letters, ensuring that the name hasn’t already been used in the names of Amber force field residues.
The residue is capped using NME and ACE groups, which has a purpose beyond maintaining the unit charge of the amino acid. When a CH3 is bonded to the pre-tail (C=O) or post-head (NH) atoms, Antechamber will recognize the C atom as an SP3 alpha-carbon and the Hs as alpha hydrogens. So, by using these capping groups, Antechamber will create the parameters for the backbone of our L-tert-Leucine (angle and dihedral values). This way, the residue will automatically bond to the previous and next residues in the chain. Please continue reading for further details.
II) Geometry Optimization
To acquire the parameters for our custom residue, its geometry needs to be optimized. This optimized geometry will be used to compute the electrostatic potential of the system. This procedure will be conducted in vacuo using B3LYP and a 6-31G* basis set.
B3LYP geometry optimization input
Submit the Calculation as usual:
g16 LTL_opt.com
Examine the log file (LTL_opt.log) in GaussView, or your preferred software, to confirm that everything is in order..
III) Electrostatic potential Charges calculation (ESP-charges)
Now, with the system in its optimized geometry, a Hartree-Fock (HF) calculation must be performed to obtain the Electrostatic Potential.
The procedure used in the development of the Amber force field to acquire the partial atom charges for every residue will be employed here. In other words, the electrostatic potential will be obtained in the gas phase using a HF/6-31G* QM calculation because “Conveniently the error in the HF/6-31G* calculation is close to the difference between the charge distribution in gas phase and that in solution “
Remember, do not change the QM level of theory when deriving charges. Use the same QM level that was used to derive the charges in the Amber forcefield.
Submit:
g16 LTL_hf.com
Output: LTL_hf.log
IV) Restrained Fitting of the Partial Charges to the Electrostatic Potentials (RESP Calculation)
The electrostatic potential must be in a format that the resp program can understand. Run the following command to extract the electrostatic potential from the Gaussian output using espgen:
submit:
espgen -i LTL_hf.log -o esp.dat
Output: esp.dat
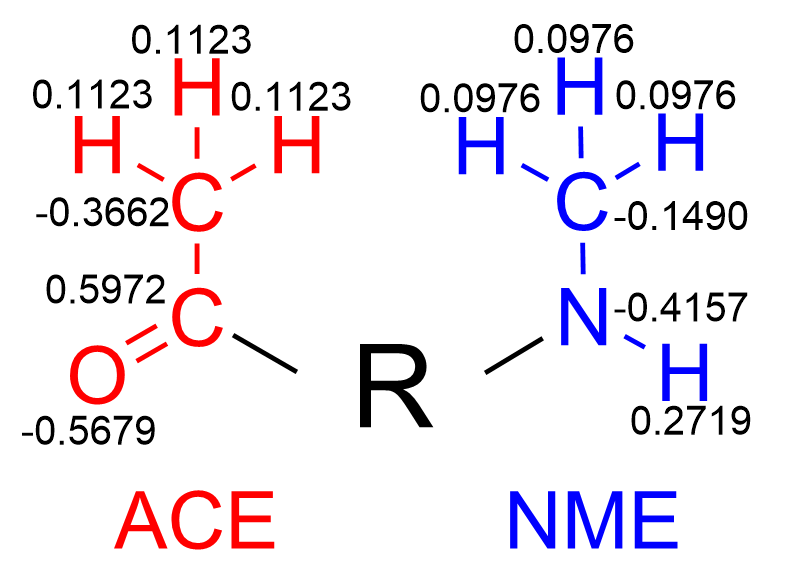
Now, we need to derive the partial charges for every atom in our residue, fitted to the ESP. Additionally, we must fix the values of the partial charges of the atoms in the ACE and NME residues to their corresponding values in the Amber force field. (Fig. 2)
Once you have the esp.dat file, you will need two additional files to instruct the RESP program which atoms will maintain a given partial charge value, and for which ones the charge will be calculated.
The first is the resp.in file. This serves as the input for this calculation (the file here contains some comments that will help you to understand it better). The description of this file isn’t included in the Amber manual, but you can read about it here: : http://ambermd.org/tutorials/advanced/tutorial1/section1.htm
resp.in (with comments, delete them before using the file)
The Second file is the resp.qin file. This is where the charges of the NME and ACE are provided to the RESP calculation.
This file has the following characteristics: each value is a partial charge, each line contains 8 entries (first line corresponds to atoms 1 to 8, the second line to atoms 9 to 16, and so on), with 10 characters for each entry. A new value is read subsequently. Each value here corresponds to an atom in the resp.in file. Thus, if a value here is different from 0.0000000, a -1 should be written in the second column for that atom in the resp.in file. Comparing the resp.in and the resp.qin files can enhance your understanding.
submit:
resp -O -i resp.in -o resp.out -p resp.pch -t resp.chg\
-q resp.qin -e esp.dat
Output: resp.out, resp.pch, resp.chg
If you encounter the “At line 403 of file resp.F (unit = 5, file = ‘resp.in’)” error, you may need to update your AmberTools. This issue was resolved in Amber 18 but not in Amber 16. Another possible error is related to the end-of-file during read. In such a case, include a blank line at the end of the resp.in file.
Another common error eported in the comments of this tutorial is:
At line 386 of file resp.F (unit = 5, file = ‘resp.in’)
Fortran runtime error: Bad value during floating point read
As Adrian Garcia pointed out in his response, the “number of the molecule” in the resp.in file is read as a floating point, while it’s written as an integer in the file. For him, changing 1 to 1.0000 solved the problem. Thanks for sharing your solutions to this issue.
In the resp.chg file are now the partial charges for each atom in the custom residue.
V) .ac File Generation or Starting to Speak Amber Language
Partial charge assignation
Here, we’ll associate the calculated partial charges with each atom in the previously optimized structure. We’ll also assign a name to the residue using Amber’s nomenclature. This is all stored in a single file, LTL.ac.
If your structure is coming from a PDB structure and you want to keep consistency with PDB naming schemes use the pdb file as input file (-i) to assign the fitted charges (-cf) to the structure using the following command:
antechamber -fi pdb -i LTL.pdb -bk LTL -fo ac\
-o LTL.ac -c rc -cf resp.chg -nc 0 -at amber
If you don’t have any pdb structure with naming scheme to conserve, you can use as input file the gaussian output:
antechamber -fi gout -i LTL_hf.log -bk LTL -fo ac\
-o LTL.ac -c rc -cf resp.chg -nc 0 -at amber
output : LTL.ac
VI) Creating the Residue for Amber Recognition
Now, we have to remove the capping groups from the custom residue and turn it into a new residue using prepgen. The main chain file contains the information needed for this. LTL.mc
The names used in the main chain file (.mc) are the same as those in the .ac file.
prepgen -i LTL.ac -o LTL.prepin -m LTL.mc -rn LTL
The final step is renaming the atom names in the .prepin file to match the atom names in the PDB file, if for some reason they differ.
output: LTL.prepin
In the 4-Hydroxyl-Proline (PR4) tutorial, the final step simply involves loading the .prepin file to obtain a system ready for simulation. However, if you’ve modified the backbone or are attempting to model a highly modified N or C terminal, prepgen may not generate the appropriate atom types for these unique atoms. Thus, you’ll need to generate a file with the atom types in the GAFF AND another with the atom types in the ff14SB. The next section demonstrates how to accomplish this when necessary. For a “standard custom residue”, we’re done.
VI.2) Generating the Parameters Files for Special Cases
By “special cases”, I refer to residues that possess a bond, angle, dihedral, or functional group not present in the protein Amber force field. As such, these parameters must be obtained using the General Amber Force Field (GAFF). To obtain them, execute:
parmchk2 -i LTL.ac -f ac -o LTL_gaff.frcmod
This command yields the file: LTL_gaff.frcmod, which needs to be included in the next step before loading the “LTL_ff14SB.frcmod” parameters. This ensures that the ff14SB parameters take precedence over GAFF whenever applicable..
You’ll know if you need to perform this step if, after completing the following step, you see lines with the message “ATTN: needs revision”:
parmchk2 -i LTL.ac -f ac -o LTL_ff14SB.frcmod -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
VII) Finally, Putting It All Together
open tleap by typing: tleap
once inside, build a test system typing the following lines
loadamberprep LTL.prepin source leaprc.protein.ff14SB
loadamberparams LTL_gaff.frcmod !if you obtained lines with the ATTN message in the ff14SB file
loadamberparams LTL_ff14SB.frcmod !after having deleted the lines with the ATTN message aa = sequence {ACE LTL NME} saveAmberParm aa aa.prmtop aa.rst7
If everything is OK, now the system is ready to run the simulation. If this isn’t the case, please leave a comment to improve this tutorial.
If you need help with a specific topic, let me know in the comments.
Recent Comments