L-tert-Leucine
This is a tutorial for intermediate level users. To comprehend it, you will need to know how to:
- Build a system to run molecular dynamics (MD) with Amber.
- Submit calculations using Gaussian.
This tutorial was designed as an alternative (or as a supplement) to the official Amber tutorials for parameterizing a custom residue. It summarizes information from three Amber tutorials:
I invested a considerable amount of time following Amber’s recipes to parameterize custom residues. I achieved my goal by combining parts of them to create my own protocol. I hope it also helps you. As I mentioned, this is a recipe; there is no technical or theoretical information included. That part is well explained in the Amber tutorials.
This is not the only or the best method, but it is one that works.
This tutorial can be viewed from a bird’s eye perspective by advanced users who are just looking for a few lines or commands to remember (or to tweak something in their regular workflow). Alternatively, beginners can follow along at a slower pace by reading the basics of what is being done. If additional information is needed, links to explanations will be included.
I) Residue Structure Selection
To calculate the partial atomic charges, we will need a 3D representation of our non-standard residue. For this, we can:
- Build it using, Gaussview, Maestro or any other software you are familiar with.
- Obtain the atomic coordinates from a PDB file or any other source that includes 3D information about the molecule (This option is highly recommended. It helps to maintain consistency with PDB naming schemes, and it makes your parameters easier for other people to use and understand).
In this case, the residue to be parameterized is L-tert-Leucine, which I refer to as LTL. However, the side chain can be anything you desire.
Keep in mind:
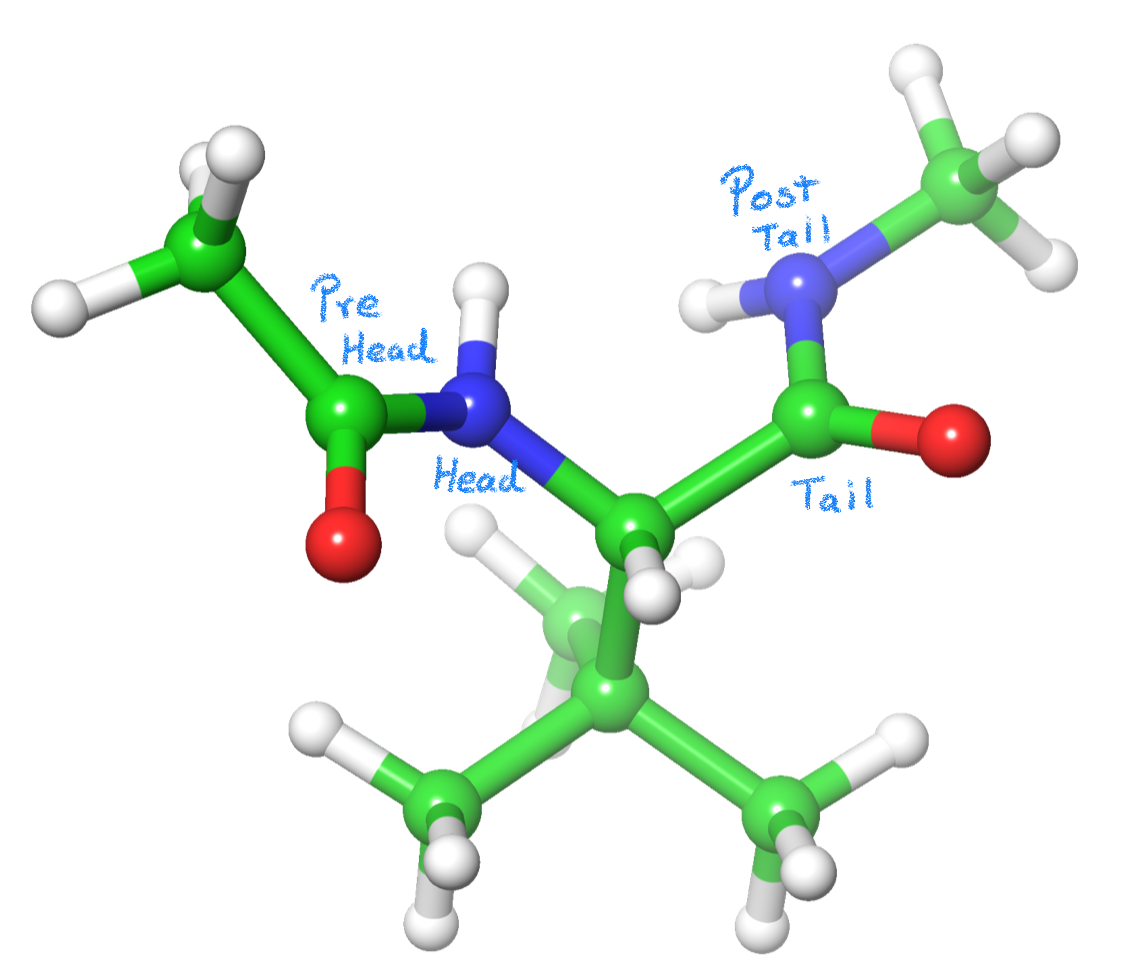
In a amino acid C=O is the tail and the NH is the head. (Fig. 1)
When naming your custom residues, use three or four capital letters, ensuring that the name hasn’t already been used in the names of Amber force field residues.
The residue is capped using NME and ACE groups, which has a purpose beyond maintaining the unit charge of the amino acid. When a CH3 is bonded to the pre-tail (C=O) or post-head (NH) atoms, Antechamber will recognize the C atom as an SP3 alpha-carbon and the Hs as alpha hydrogens. So, by using these capping groups, Antechamber will create the parameters for the backbone of our L-tert-Leucine (angle and dihedral values). This way, the residue will automatically bond to the previous and next residues in the chain. Please continue reading for further details.
II) Geometry Optimization
To acquire the parameters for our custom residue, its geometry needs to be optimized. This optimized geometry will be used to compute the electrostatic potential of the system. This procedure will be conducted in vacuo using B3LYP and a 6-31G* basis set.
B3LYP geometry optimization input
Submit the Calculation as usual:
g16 LTL_opt.com
Examine the log file (LTL_opt.log) in GaussView, or your preferred software, to confirm that everything is in order..
III) Electrostatic potential Charges calculation (ESP-charges)
Now, with the system in its optimized geometry, a Hartree-Fock (HF) calculation must be performed to obtain the Electrostatic Potential.
The procedure used in the development of the Amber force field to acquire the partial atom charges for every residue will be employed here. In other words, the electrostatic potential will be obtained in the gas phase using a HF/6-31G* QM calculation because “Conveniently the error in the HF/6-31G* calculation is close to the difference between the charge distribution in gas phase and that in solution “
Remember, do not change the QM level of theory when deriving charges. Use the same QM level that was used to derive the charges in the Amber forcefield.
Submit:
g16 LTL_hf.com
Output: LTL_hf.log
IV) Restrained Fitting of the Partial Charges to the Electrostatic Potentials (RESP Calculation)
The electrostatic potential must be in a format that the resp program can understand. Run the following command to extract the electrostatic potential from the Gaussian output using espgen:
submit:
espgen -i LTL_hf.log -o esp.dat
Output: esp.dat
Now, we need to derive the partial charges for every atom in our residue, fitted to the ESP. Additionally, we must fix the values of the partial charges of the atoms in the ACE and NME residues to their corresponding values in the Amber force field. (Fig. 2)
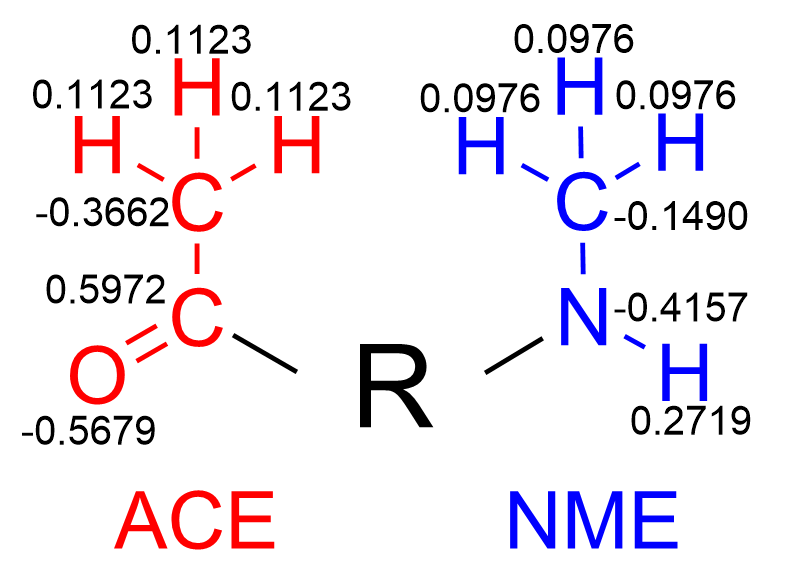
Once you have the esp.dat file, you will need two additional files to instruct the RESP program which atoms will maintain a given partial charge value, and for which ones the charge will be calculated.
The first is the resp.in file. This serves as the input for this calculation (the file here contains some comments that will help you to understand it better). The description of this file isn’t included in the Amber manual, but you can read about it here: : http://ambermd.org/tutorials/advanced/tutorial1/section1.htm
resp.in (with comments, delete them before using the file)
The Second file is the resp.qin file. This is where the charges of the NME and ACE are provided to the RESP calculation.
This file has the following characteristics: each value is a partial charge, each line contains 8 entries (first line corresponds to atoms 1 to 8, the second line to atoms 9 to 16, and so on), with 10 characters for each entry. A new value is read subsequently. Each value here corresponds to an atom in the resp.in file. Thus, if a value here is different from 0.0000000, a -1 should be written in the second column for that atom in the resp.in file. Comparing the resp.in and the resp.qin files can enhance your understanding.
submit:
resp -O -i resp.in -o resp.out -p resp.pch -t resp.chg\
-q resp.qin -e esp.dat
Output: resp.out, resp.pch, resp.chg
If you encounter the “At line 403 of file resp.F (unit = 5, file = ‘resp.in’)” error, you may need to update your AmberTools. This issue was resolved in Amber 18 but not in Amber 16. Another possible error is related to the end-of-file during read. In such a case, include a blank line at the end of the resp.in file.
Another common error eported in the comments of this tutorial is:
At line 386 of file resp.F (unit = 5, file = ‘resp.in’)
Fortran runtime error: Bad value during floating point read
As Adrian Garcia pointed out in his response, the “number of the molecule” in the resp.in file is read as a floating point, while it’s written as an integer in the file. For him, changing 1 to 1.0000 solved the problem. Thanks for sharing your solutions to this issue.
In the resp.chg file are now the partial charges for each atom in the custom residue.
V) .ac File Generation or Starting to Speak Amber Language
Partial charge assignation
Here, we’ll associate the calculated partial charges with each atom in the previously optimized structure. We’ll also assign a name to the residue using Amber’s nomenclature. This is all stored in a single file, LTL.ac.
If your structure is coming from a PDB structure and you want to keep consistency with PDB naming schemes use the pdb file as input file (-i) to assign the fitted charges (-cf) to the structure using the following command:
antechamber -fi pdb -i LTL.pdb -bk LTL -fo ac\
-o LTL.ac -c rc -cf resp.chg -nc 0 -at amber
If you don’t have any pdb structure with naming scheme to conserve, you can use as input file the gaussian output:
antechamber -fi gout -i LTL_hf.log -bk LTL -fo ac\
-o LTL.ac -c rc -cf resp.chg -nc 0 -at amber
output : LTL.ac
VI) Creating the Residue for Amber Recognition
Now, we have to remove the capping groups from the custom residue and turn it into a new residue using prepgen. The main chain file contains the information needed for this. LTL.mc
The names used in the main chain file (.mc) are the same as those in the .ac file.
prepgen -i LTL.ac -o LTL.prepin -m LTL.mc -rn LTL
The final step is renaming the atom names in the .prepin file to match the atom names in the PDB file, if for some reason they differ.
output: LTL.prepin
In the 4-Hydroxyl-Proline (PR4) tutorial, the final step simply involves loading the .prepin file to obtain a system ready for simulation. However, if you’ve modified the backbone or are attempting to model a highly modified N or C terminal, prepgen may not generate the appropriate atom types for these unique atoms. Thus, you’ll need to generate a file with the atom types in the GAFF AND another with the atom types in the ff14SB. The next section demonstrates how to accomplish this when necessary. For a “standard custom residue”, we’re done.
VI.2) Generating the Parameters Files for Special Cases
By “special cases”, I refer to residues that possess a bond, angle, dihedral, or functional group not present in the protein Amber force field. As such, these parameters must be obtained using the General Amber Force Field (GAFF). To obtain them, execute:
parmchk2 -i LTL.ac -f ac -o LTL_gaff.frcmod
This command yields the file: LTL_gaff.frcmod, which needs to be included in the next step before loading the “LTL_ff14SB.frcmod” parameters. This ensures that the ff14SB parameters take precedence over GAFF whenever applicable..
You’ll know if you need to perform this step if, after completing the following step, you see lines with the message “ATTN: needs revision”:
parmchk2 -i LTL.ac -f ac -o LTL_ff14SB.frcmod -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
VII) Finally, Putting It All Together
open tleap by typing: tleap
once inside, build a test system typing the following lines
loadamberprep LTL.prepin source leaprc.protein.ff14SB
loadamberparams LTL_gaff.frcmod !if you obtained lines with the ATTN message in the ff14SB file
loadamberparams LTL_ff14SB.frcmod !after having deleted the lines with the ATTN message aa = sequence {ACE LTL NME} saveAmberParm aa aa.prmtop aa.rst7
If everything is OK, now the system is ready to run the simulation. If this isn’t the case, please leave a comment to improve this tutorial.
If you need help with a specific topic, let me know in the comments.
Dear,
I’m trying your tutorial and got stuck in the following error:
At line 386 of file resp.F (unit = 5, file = ‘resp.in’)
Fortran runtime error: Bad value during floating point read
Could you please give me any advice?
Thank you!
Hi Anderson.
Did you delete the text after the exclamation signs in the resp.in file? ah! and don’t forget to include a blank line after the last line in the text.
try again and let me know if that was the problem.
Regards.
I had a similar problem and by looking at the code in resp.F I noticed that the variable reading “number of the molecule” in the resp.in file is defined as floating point when read, whereas in the file is written as an integer. Changuing 1 by 1.0000 solved the problem for me, so try that.
Did this tutorial without having ever worked with amber before. It was easy to follow and everything worked out perfectly fine. Great tutorial- Thank you for your work!
Thank you so much for preparing such a great tutorial on “Amber Custom Residue Parameterization”. I got an issue at RESP step calculation for my model which is ligand covalently bonded to Cys residue of protein. (resp -O -i resp.in -o resp.out -p resp.pch -t resp.chg -q resp.qin -e esp.dat). It will be great to have an email for sharing results with you which is a big help to me fixing the issue. I appreciate your consideration.
Bests
Zahra
Hello Zahra,
Thank you for your interest in this tutorial. If you wish to parameterize a residue bonded to a Cys residue, likely through the sulfur, the recommended approach is to begin with an initial structure that includes both residues bonded to each other. Additionally, ensure that each residue has its corresponding cap termini residues ACE and NME.
your initial structure should looks like this:
If you follow this approach, you can parameterize your custom Cysteine (CYA) and your custom residue (RES). To obtain charges for each atom in the RESP program, ensure the preparation of corresponding resp.in and resp.qin files, incorporating charge values for atoms in the ACE and NME residues, as demonstrated in the tutorial. If you choose not to parameterise the new Cys bonded to your RES, include the charges of the CYX amber residue in the resp.qin file. This way, only the charges of RES will be fitted based on the values provided in the resp.qin file.
The main distinction arises when creating the unit without additional atoms, particularly when creating the residue for Amber recognition. Specify the exclusion of atoms by indicating their numbers in the main chain file. Your RES.mc file should resemble the following:
HEAD_NAME N1
TAIL_NAME C8
MAIN_CHAIN C3
OMIT_NAME C1
.
.
.
OMIT_NAME H18
PRE_HEAD_TYPE C
POST_TAIL_TYPE N
CHARGE 0.0 (the charge of your residue)
Adapt it accordingly to your system. List each atom you wish to exclude from your residue by employing the OMIT_NAME keyword, as exemplified in the LTL.mc example file.
Proceed with the rest of the tutorial to generate the frcmod and prepin files.
When preparing your system using leap, remember to bond the two atoms between CYS and RES with the following line:
bond SYSTEM.XX.AA SYSTEM.YY.BB
Here, SYSTEM is the name you assign to your system in leap, XX denotes the number of the Cys atom named AA bonded to the atom named BB with number YY in your “RES.”
I hope this information helps you with the parameterization of your custom residue.
Great! Thank you so much Carlos for the prompt response and sharing technical details. I appreciate that. I will follow all details you mentioned. Yes, there is a covalent bond between S-Cys and C-ligand so I am wondering if I need to CAP ligand or not, and the ligand has charge of -2 at its terminal group. I have considered this coordination model to start optimization with Gaussian, If it looks fine then I can move on with other steps.
Bests
Zahra
C -2.69100000 -17.32800000 23.67100000
C -3.97800000 -16.74700000 24.26500000
O -4.15700000 -16.81200000 25.49900000
H -2.69500000 -17.18900000 22.59000000
H -2.63300000 -18.39200000 23.90000000
H -1.82900000 -16.81600000 24.10000000
N -4.84000000 -16.19000000 23.41000000
C -6.12000000 -15.51000000 23.79000000
C -6.86000000 -15.13000000 22.50000000
S -8.23000000 -13.97000000 22.87000000
C -6.96000000 -16.39000000 24.74000000
O -7.67000000 -15.81000000 25.59000000
C -9.79000000 -14.58000000 21.84000000
C -10.90000000 -14.33000000 22.87000000
N -10.02000000 -13.80000000 20.68000000
P -8.47000000 -23.96000000 22.39000000
O -9.80000000 -24.68000000 22.12000000
O -7.17000000 -24.74000000 22.02000000
O -8.36000000 -23.29000000 23.80000000
C -9.55000000 -22.01000000 20.83000000
O -8.37000000 -22.66000000 21.27000000
C -9.43000000 -20.49000000 20.94000000
O -9.67000000 -20.08000000 22.29000000
C -8.05000000 -19.99000000 20.49000000
O -7.88000000 -20.28000000 19.10000000
C -7.84000000 -18.48000000 20.71000000
O -6.53000000 -18.11000000 20.28000000
C -8.85000000 -17.68000000 19.90000000
N -10.06000000 -17.02000000 22.24000000
C -10.81000000 -16.77000000 23.36000000
O -11.16000000 -17.65000000 24.15000000
N -11.25000000 -15.44000000 23.59000000
O -11.38000000 -13.22000000 23.03000000
C -9.06000000 -13.94000000 19.68000000
C -8.67000000 -12.90000000 18.82000000
C -7.69000000 -13.09000000 17.84000000
C -7.29000000 -11.95000000 16.94000000
C -7.06000000 -14.36000000 17.72000000
C -5.97000000 -14.59000000 16.71000000
C -7.46000000 -15.40000000 18.57000000
C -8.47000000 -15.21000000 19.53000000
C -9.63000000 -16.02000000 21.49000000
N -8.96000000 -16.27000000 20.34000000
H -4.61000000 -16.19000000 22.42000000
H -5.88000000 -14.60000000 24.37000000
H -7.23000000 -16.03000000 21.99000000
H -6.16000000 -14.62000000 21.81000000
H -10.43000000 -22.34000000 21.40000000
H -9.72000000 -22.26000000 19.76000000
H -10.21000000 -20.07000000 20.26000000
H -9.84000000 -19.10000000 22.29000000
H -7.30000000 -20.53000000 21.09000000
H -7.05000000 -19.82000000 18.85000000
H -7.96000000 -18.26000000 21.78000000
H -5.92000000 -18.42000000 21.00000000
H -9.86000000 -18.11000000 20.00000000
H -8.58000000 -17.68000000 18.84000000
H -11.90000000 -15.31000000 24.37000000
H -10.38000000 -12.86000000 20.87000000
H -9.14000000 -11.91000000 18.94000000
H -6.21000000 -11.71000000 17.05000000
H -7.44000000 -12.21000000 15.87000000
H -7.87000000 -11.04000000 17.16000000
H -5.11000000 -13.91000000 16.87000000
H -5.59000000 -15.63000000 16.76000000
H -6.33000000 -14.40000000 15.68000000
H -6.96000000 -16.38000000 18.51000000
N -6.85500000 -17.72100000 24.64600000
C -7.80200000 -18.61800000 25.35500000
H -6.11900000 -18.12200000 24.08300000
H -8.52400000 -18.01900000 25.90900000
H -7.25100000 -19.25500000 26.04700000
H -8.32700000 -19.23900000 24.62900000
Hi Zahra,
Having visualised your structure, it’s clear to me that you don’t need to cap your “RES” residue as it’s unlikely to be attached to a backbone chain. If this is the case, you can start optimising the geometry of this structure and then follow the tutorial.
Let me know if this is clear enough.
Hi Carlos. I have done geom optimization, RESP (-2 charge, 73 atoms and setting -1 to ACE and NME residues and other atoms equal to zero). There were two issues at RESP steps (FORTRAN runtime error: Bad value during floating point read, Fortran runtime error: End of file) and have been fixed based on reported commands from other users. Then, antechamber -fi gout -i FMN-hf.log -bk FMN -fo ac -o FMN.ac -c rc -cf resp.chg -at amber, here is the FMN.ac file:
(base) cat FMN.ac
CHARGE -2.00 ( -2 )
Formula: H31 C23 N6 O11 P1 S1
ATOM 1 N1 MOL 1 -1.402 -1.545 3.066 -0.591590 N
ATOM 2 N2 MOL 1 -0.334 -3.052 0.932 -0.342128 N
ATOM 3 C1 MOL 1 -2.278 -2.023 2.002 -0.025870 CT
ATOM 4 C2 MOL 1 -0.100 -1.751 5.125 -0.612845 CT
ATOM 5 C3 MOL 1 0.415 -4.051 0.182 -0.334400 CT
ATOM 6 C4 MOL 1 -1.652 -3.191 1.205 0.660925 C
ATOM 7 C5 MOL 1 -1.043 -2.359 4.104 0.783892 C
ATOM 8 O1 MOL 1 -2.337 -4.141 0.808 -0.612160 O
ATOM 9 O2 MOL 1 -1.451 -3.526 4.174 -0.611120 O
ATOM 10 C6 MOL 1 -2.576 -0.818 1.089 -0.543266 CT
ATOM 11 S1 MOL 1 -3.612 -1.308 -0.343 0.000000 S
ATOM 12 P1 MOL 1 6.748 0.192 -0.040 0.000000 P
ATOM 13 O3 MOL 1 7.293 -0.106 -1.453 -0.544005 O2
ATOM 14 O4 MOL 1 7.606 1.139 0.833 -0.610434 O2
ATOM 15 O5 MOL 1 6.171 -1.023 0.735 -0.574457 O2
ATOM 16 C7 MOL 1 4.666 1.277 -1.581 0.044452 CT
ATOM 17 O6 MOL 1 5.386 1.204 -0.401 -0.175829 OS
ATOM 18 C8 MOL 1 3.337 0.530 -1.462 -0.022976 CT
ATOM 19 O7 MOL 1 3.659 -0.851 -1.651 -0.496317 OH
ATOM 20 C9 MOL 1 2.675 0.736 -0.100 0.131863 CT
ATOM 21 O8 MOL 1 2.771 2.117 0.218 -0.656784 OH
ATOM 22 C10 MOL 1 1.208 0.295 -0.082 0.045337 CT
ATOM 23 O9 MOL 1 0.658 0.441 1.204 -0.608289 OH
ATOM 24 C11 MOL 1 0.466 1.144 -1.081 -0.072047 CT
ATOM 25 N3 MOL 1 -0.881 -0.765 -2.454 -0.492301 DU
ATOM 26 C12 MOL 1 -1.475 -1.698 -3.268 0.595808 C
ATOM 27 O10 MOL 1 -0.863 -2.626 -3.792 -0.539308 O
ATOM 28 N4 MOL 1 -2.858 -1.540 -3.563 -0.378562 N
ATOM 29 C13 MOL 1 -3.714 -0.672 -2.946 0.540779 C
ATOM 30 C14 MOL 1 -3.119 0.021 -1.716 -0.347934 CT
ATOM 31 O11 MOL 1 -4.879 -0.481 -3.279 -0.517951 O
ATOM 32 C15 MOL 1 -3.144 2.153 -0.608 0.007567 CA
ATOM 33 N5 MOL 1 -3.692 1.299 -1.557 -0.296909 NT
ATOM 34 C16 MOL 1 -3.898 3.148 0.037 -0.271554 CA
ATOM 35 C17 MOL 1 -3.317 4.014 0.974 0.070769 CA
ATOM 36 C18 MOL 1 -4.155 5.071 1.653 -0.241331 CT
ATOM 37 C19 MOL 1 -1.937 3.871 1.286 0.072834 CA
ATOM 38 C20 MOL 1 -1.277 4.766 2.309 -0.240244 CT
ATOM 39 C21 MOL 1 -1.185 2.881 0.636 -0.217269 CA
ATOM 40 C22 MOL 1 -1.765 2.027 -0.318 0.017070 CA
ATOM 41 C23 MOL 1 -1.616 0.084 -1.767 0.693489 CM
ATOM 42 N6 MOL 1 -1.011 1.053 -1.027 -0.074009 NT
ATOM 43 H1 MOL 1 0.338 -5.041 0.668 0.140227 H1
ATOM 44 H2 MOL 1 1.470 -3.742 0.145 0.153090 H1
ATOM 45 H3 MOL 1 -0.496 -1.942 6.136 0.174640 HC
ATOM 46 H4 MOL 1 0.876 -2.262 5.047 0.190410 HC
ATOM 47 H5 MOL 1 -1.058 -0.584 3.031 0.331452 H
ATOM 48 H6 MOL 1 0.165 -2.275 1.363 0.292543 H
ATOM 49 H7 MOL 1 0.049 -0.670 4.981 0.154416 HC
ATOM 50 H8 MOL 1 -3.208 -2.429 2.434 0.178143 H1
ATOM 51 H9 MOL 1 0.028 -4.134 -0.849 0.146485 H1
ATOM 52 H10 MOL 1 -1.614 -0.420 0.728 0.173219 H1
ATOM 53 H11 MOL 1 -3.091 -0.033 1.668 0.263073 H1
ATOM 54 H12 MOL 1 5.229 0.827 -2.426 0.063424 H1
ATOM 55 H13 MOL 1 4.453 2.339 -1.827 0.016626 H1
ATOM 56 H14 MOL 1 2.679 0.882 -2.282 0.111883 H1
ATOM 57 H15 MOL 1 2.826 -1.352 -1.742 0.318709 HO
ATOM 58 H16 MOL 1 3.254 0.121 0.620 0.124128 H1
ATOM 59 H17 MOL 1 2.341 2.236 1.088 0.422025 HO
ATOM 60 H18 MOL 1 1.149 -0.766 -0.401 0.090879 H1
ATOM 61 H19 MOL 1 1.198 -0.083 1.828 0.412515 HO
ATOM 62 H20 MOL 1 0.747 0.830 -2.093 0.009937 H1
ATOM 63 H21 MOL 1 0.730 2.201 -0.947 0.148513 H1
ATOM 64 H22 MOL 1 -3.235 -2.133 -4.307 0.309656 H
ATOM 65 H23 MOL 1 -4.699 1.339 -1.727 0.331129 H
ATOM 66 H24 MOL 1 -4.963 3.234 -0.205 0.180659 HA
ATOM 67 H25 MOL 1 -4.164 4.930 2.750 0.076636 HC
ATOM 68 H26 MOL 1 -3.749 6.082 1.467 0.084836 HC
ATOM 69 H27 MOL 1 -5.196 5.044 1.293 0.065213 HC
ATOM 70 H28 MOL 1 -1.762 4.673 3.298 0.066900 HC
ATOM 71 H29 MOL 1 -0.210 4.515 2.423 0.084432 HC
ATOM 72 H30 MOL 1 -1.352 5.831 2.019 0.076821 HC
ATOM 73 H31 MOL 1 -0.134 2.758 0.897 0.194484 HA
BOND 1 1 3 1 N1 C1
BOND 2 1 7 1 N1 C5
BOND 3 1 47 1 N1 H5
BOND 4 2 5 1 N2 C3
BOND 5 2 6 1 N2 C4
BOND 6 2 48 1 N2 H6
BOND 7 3 6 1 C1 C4
BOND 8 3 10 1 C1 C6
BOND 9 3 50 1 C1 H8
BOND 10 4 7 1 C2 C5
BOND 11 4 45 1 C2 H3
BOND 12 4 46 1 C2 H4
BOND 13 4 49 1 C2 H7
BOND 14 5 43 1 C3 H1
BOND 15 5 44 1 C3 H2
BOND 16 5 51 1 C3 H9
BOND 17 6 8 2 C4 O1
BOND 18 7 9 2 C5 O2
BOND 19 10 11 1 C6 S1
BOND 20 10 52 1 C6 H10
BOND 21 10 53 1 C6 H11
BOND 22 11 30 1 S1 C14
BOND 23 12 13 9 P1 O3
BOND 24 12 14 9 P1 O4
BOND 25 12 15 9 P1 O5
BOND 26 12 17 1 P1 O6
BOND 27 16 17 1 C7 O6
BOND 28 16 18 1 C7 C8
BOND 29 16 54 1 C7 H12
BOND 30 16 55 1 C7 H13
BOND 31 18 19 1 C8 O7
BOND 32 18 20 1 C8 C9
BOND 33 18 56 1 C8 H14
BOND 34 19 57 1 O7 H15
BOND 35 20 21 1 C9 O8
BOND 36 20 22 1 C9 C10
BOND 37 20 58 1 C9 H16
BOND 38 21 59 1 O8 H17
BOND 39 22 23 1 C10 O9
BOND 40 22 24 1 C10 C11
BOND 41 22 60 1 C10 H18
BOND 42 23 61 1 O9 H19
BOND 43 24 42 1 C11 N6
BOND 44 24 62 1 C11 H20
BOND 45 24 63 1 C11 H21
BOND 46 25 26 1 N3 C12
BOND 47 25 41 2 N3 C23
BOND 48 26 27 2 C12 O10
BOND 49 26 28 1 C12 N4
BOND 50 28 29 1 N4 C13
BOND 51 28 64 1 N4 H22
BOND 52 29 30 1 C13 C14
BOND 53 29 31 2 C13 O11
BOND 54 30 33 1 C14 N5
BOND 55 30 41 1 C14 C23
BOND 56 32 33 1 C15 N5
BOND 57 32 34 7 C15 C16
BOND 58 32 40 8 C15 C22
BOND 59 33 65 1 N5 H23
BOND 60 34 35 8 C16 C17
BOND 61 34 66 1 C16 H24
BOND 62 35 36 1 C17 C18
BOND 63 35 37 7 C17 C19
BOND 64 36 67 1 C18 H25
BOND 65 36 68 1 C18 H26
BOND 66 36 69 1 C18 H27
BOND 67 37 38 1 C19 C20
BOND 68 37 39 8 C19 C21
BOND 69 38 70 1 C20 H28
BOND 70 38 71 1 C20 H29
BOND 71 38 72 1 C20 H30
BOND 72 39 40 7 C21 C22
BOND 73 39 73 1 C21 H31
BOND 74 40 42 1 C22 N6
BOND 75 41 42 1 C23 N6
Ok, this is good. But remember that if you do it this way, you will end up with one large residue containing both your “RES” and the CYS to which it is attached. This means that your “FMN” unit is actually two residues and that they need to be included in your PDB file in this way. Before continuing with the parameterisation process, let me know if this is what you want to do, because if it is not, the approach to solving the problem will be different.
I’ll be happy to help you with this.
Hi Carlos. Thank you ! yes, the residue is a bit big and I need to consider them as a merged residue with name of FMN and should also rename it on final PDB file. I think you have seen my message out of order that I posted 🙂 but it should be fine. There was an issue with parmchk2 for preparing FMN_gaff.frcmod file and it has been fixed. Now I have prepared FMN_gaff.frcmod and FMN_ff14SB.frcmod but both have couple of records tagged with “ATTN, need revision”. Can you please let me know how to modify them for using in tleap and writing parm files for MD. Bests/Zahra
Then, prepgen -i FMN.ac -o FMN.prepin -m FMN.mc -rn FMN as:
PRE_HEAD_TYPE is C
POST_TAIL_TYPE is N
Net charge of truncated molecule is -2.00
HEAD_ATOM 1 N1
TAIL_ATOM 6 C4
MAIN_CHAIN 1 1 N1
MAIN_CHAIN 2 3 C1
MAIN_CHAIN 3 6 C4
OMIT_ATOM 1 4 C2
OMIT_ATOM 2 45 H3
OMIT_ATOM 3 46 H4
OMIT_ATOM 4 49 H7
OMIT_ATOM 5 7 C5
OMIT_ATOM 6 9 O2
OMIT_ATOM 7 2 N2
OMIT_ATOM 8 48 H6
OMIT_ATOM 9 5 C3
OMIT_ATOM 10 43 H1
OMIT_ATOM 11 44 H2
OMIT_ATOM 12 51 H9
Number of mainchain atoms (including head and tail atom): 3
Number of omited atoms: 12
Warning: ATOM N3 has unfilled valence, assuming the linked atom name (in other residue) is “M”
change “M” to “-M” if ATOM N3 is linked to the immediate previous residue
change “M” to “+M” if ATOM N3 is linked to the immediate after residue(base)
then I stopped at the following step with Segmentation fault error.
parmchk2 -i FMN.ac -f ac -o FMN_gaff.frcmod
Warning: Atom type (DU) is not in PARMCHK.DAT; using default values
for improper_flag [0], group_id [0], and equivalent_flag [0].
Warning: No mass information for atom type (DU); set to 0.0.
It is recommended to add the new atom type (DU) to PARMCHK.DAT.
/home/jirka/amber18/bin/parmchk2: line 4: 65155 Segmentation fault (core dumped) $AMBERHOME/bin/to_be_dispatched/parmchk2 “$@”
Hi Zahra, could you please repeat the generation of the .ac file with antechamber but this time use ‘-at gaff2’ instead of ‘-at amber’. As your molecule is very different from a protein or a nucleic acid it is likely that this is the reason why amber is not recognising your N3 nitrogen and is assigning the DU atom type.
Please let me know if this solves the problem.
Sure! Actually it is combination of amino acid + organic molecule, here is the .ac file using -at gaff2 :
(base) cat FMN.ac
CHARGE -2.00 ( -2 )
Formula: H31 C23 N6 O11 P1 S1
ATOM 1 N1 MOL 1 -1.402 -1.545 3.066 -0.591590 ns
ATOM 2 N2 MOL 1 -0.334 -3.052 0.932 -0.342128 ns
ATOM 3 C1 MOL 1 -2.278 -2.023 2.002 -0.025870 c3
ATOM 4 C2 MOL 1 -0.100 -1.751 5.125 -0.612845 c3
ATOM 5 C3 MOL 1 0.415 -4.051 0.182 -0.334400 c3
ATOM 6 C4 MOL 1 -1.652 -3.191 1.205 0.660925 c
ATOM 7 C5 MOL 1 -1.043 -2.359 4.104 0.783892 c
ATOM 8 O1 MOL 1 -2.337 -4.141 0.808 -0.612160 o
ATOM 9 O2 MOL 1 -1.451 -3.526 4.174 -0.611120 o
ATOM 10 C6 MOL 1 -2.576 -0.818 1.089 -0.543266 c3
ATOM 11 S1 MOL 1 -3.612 -1.308 -0.343 0.000000 ss
ATOM 12 P1 MOL 1 6.748 0.192 -0.040 0.000000 p5
ATOM 13 O3 MOL 1 7.293 -0.106 -1.453 -0.544005 o
ATOM 14 O4 MOL 1 7.606 1.139 0.833 -0.610434 o
ATOM 15 O5 MOL 1 6.171 -1.023 0.735 -0.574457 o
ATOM 16 C7 MOL 1 4.666 1.277 -1.581 0.044452 c3
ATOM 17 O6 MOL 1 5.386 1.204 -0.401 -0.175829 os
ATOM 18 C8 MOL 1 3.337 0.530 -1.462 -0.022976 c3
ATOM 19 O7 MOL 1 3.659 -0.851 -1.651 -0.496317 oh
ATOM 20 C9 MOL 1 2.675 0.736 -0.100 0.131863 c3
ATOM 21 O8 MOL 1 2.771 2.117 0.218 -0.656784 oh
ATOM 22 C10 MOL 1 1.208 0.295 -0.082 0.045337 c3
ATOM 23 O9 MOL 1 0.658 0.441 1.204 -0.608289 oh
ATOM 24 C11 MOL 1 0.466 1.144 -1.081 -0.072047 c3
ATOM 25 N3 MOL 1 -0.881 -0.765 -2.454 -0.492301 ne
ATOM 26 C12 MOL 1 -1.475 -1.698 -3.268 0.595808 c
ATOM 27 O10 MOL 1 -0.863 -2.626 -3.792 -0.539308 o
ATOM 28 N4 MOL 1 -2.858 -1.540 -3.563 -0.378562 ns
ATOM 29 C13 MOL 1 -3.714 -0.672 -2.946 0.540779 c
ATOM 30 C14 MOL 1 -3.119 0.021 -1.716 -0.347934 c3
ATOM 31 O11 MOL 1 -4.879 -0.481 -3.279 -0.517951 o
ATOM 32 C15 MOL 1 -3.144 2.153 -0.608 0.007567 ca
ATOM 33 N5 MOL 1 -3.692 1.299 -1.557 -0.296909 nu
ATOM 34 C16 MOL 1 -3.898 3.148 0.037 -0.271554 ca
ATOM 35 C17 MOL 1 -3.317 4.014 0.974 0.070769 ca
ATOM 36 C18 MOL 1 -4.155 5.071 1.653 -0.241331 c3
ATOM 37 C19 MOL 1 -1.937 3.871 1.286 0.072834 ca
ATOM 38 C20 MOL 1 -1.277 4.766 2.309 -0.240244 c3
ATOM 39 C21 MOL 1 -1.185 2.881 0.636 -0.217269 ca
ATOM 40 C22 MOL 1 -1.765 2.027 -0.318 0.017070 ca
ATOM 41 C23 MOL 1 -1.616 0.084 -1.767 0.693489 c2
ATOM 42 N6 MOL 1 -1.011 1.053 -1.027 -0.074009 nh
ATOM 43 H1 MOL 1 0.338 -5.041 0.668 0.140227 h1
ATOM 44 H2 MOL 1 1.470 -3.742 0.145 0.153090 h1
ATOM 45 H3 MOL 1 -0.496 -1.942 6.136 0.174640 hc
ATOM 46 H4 MOL 1 0.876 -2.262 5.047 0.190410 hc
ATOM 47 H5 MOL 1 -1.058 -0.584 3.031 0.331452 hn
ATOM 48 H6 MOL 1 0.165 -2.275 1.363 0.292543 hn
ATOM 49 H7 MOL 1 0.049 -0.670 4.981 0.154416 hc
ATOM 50 H8 MOL 1 -3.208 -2.429 2.434 0.178143 h1
ATOM 51 H9 MOL 1 0.028 -4.134 -0.849 0.146485 h1
ATOM 52 H10 MOL 1 -1.614 -0.420 0.728 0.173219 h1
ATOM 53 H11 MOL 1 -3.091 -0.033 1.668 0.263073 h1
ATOM 54 H12 MOL 1 5.229 0.827 -2.426 0.063424 h1
ATOM 55 H13 MOL 1 4.453 2.339 -1.827 0.016626 h1
ATOM 56 H14 MOL 1 2.679 0.882 -2.282 0.111883 h1
ATOM 57 H15 MOL 1 2.826 -1.352 -1.742 0.318709 ho
ATOM 58 H16 MOL 1 3.254 0.121 0.620 0.124128 h1
ATOM 59 H17 MOL 1 2.341 2.236 1.088 0.422025 ho
ATOM 60 H18 MOL 1 1.149 -0.766 -0.401 0.090879 h1
ATOM 61 H19 MOL 1 1.198 -0.083 1.828 0.412515 ho
ATOM 62 H20 MOL 1 0.747 0.830 -2.093 0.009937 h1
ATOM 63 H21 MOL 1 0.730 2.201 -0.947 0.148513 h1
ATOM 64 H22 MOL 1 -3.235 -2.133 -4.307 0.309656 hn
ATOM 65 H23 MOL 1 -4.699 1.339 -1.727 0.331129 hn
ATOM 66 H24 MOL 1 -4.963 3.234 -0.205 0.180659 ha
ATOM 67 H25 MOL 1 -4.164 4.930 2.750 0.076636 hc
ATOM 68 H26 MOL 1 -3.749 6.082 1.467 0.084836 hc
ATOM 69 H27 MOL 1 -5.196 5.044 1.293 0.065213 hc
ATOM 70 H28 MOL 1 -1.762 4.673 3.298 0.066900 hc
ATOM 71 H29 MOL 1 -0.210 4.515 2.423 0.084432 hc
ATOM 72 H30 MOL 1 -1.352 5.831 2.019 0.076821 hc
ATOM 73 H31 MOL 1 -0.134 2.758 0.897 0.194484 ha
BOND 1 1 3 1 N1 C1
BOND 2 1 7 1 N1 C5
BOND 3 1 47 1 N1 H5
BOND 4 2 5 1 N2 C3
BOND 5 2 6 1 N2 C4
BOND 6 2 48 1 N2 H6
BOND 7 3 6 1 C1 C4
BOND 8 3 10 1 C1 C6
BOND 9 3 50 1 C1 H8
BOND 10 4 7 1 C2 C5
BOND 11 4 45 1 C2 H3
BOND 12 4 46 1 C2 H4
BOND 13 4 49 1 C2 H7
BOND 14 5 43 1 C3 H1
BOND 15 5 44 1 C3 H2
BOND 16 5 51 1 C3 H9
BOND 17 6 8 2 C4 O1
BOND 18 7 9 2 C5 O2
BOND 19 10 11 1 C6 S1
BOND 20 10 52 1 C6 H10
BOND 21 10 53 1 C6 H11
BOND 22 11 30 1 S1 C14
BOND 23 12 13 9 P1 O3
BOND 24 12 14 9 P1 O4
BOND 25 12 15 9 P1 O5
BOND 26 12 17 1 P1 O6
BOND 27 16 17 1 C7 O6
BOND 28 16 18 1 C7 C8
BOND 29 16 54 1 C7 H12
BOND 30 16 55 1 C7 H13
BOND 31 18 19 1 C8 O7
BOND 32 18 20 1 C8 C9
BOND 33 18 56 1 C8 H14
BOND 34 19 57 1 O7 H15
BOND 35 20 21 1 C9 O8
BOND 36 20 22 1 C9 C10
BOND 37 20 58 1 C9 H16
BOND 38 21 59 1 O8 H17
BOND 39 22 23 1 C10 O9
BOND 40 22 24 1 C10 C11
BOND 41 22 60 1 C10 H18
BOND 42 23 61 1 O9 H19
BOND 43 24 42 1 C11 N6
BOND 44 24 62 1 C11 H20
BOND 45 24 63 1 C11 H21
BOND 46 25 26 1 N3 C12
BOND 47 25 41 2 N3 C23
BOND 48 26 27 2 C12 O10
BOND 49 26 28 1 C12 N4
BOND 50 28 29 1 N4 C13
BOND 51 28 64 1 N4 H22
BOND 52 29 30 1 C13 C14
BOND 53 29 31 2 C13 O11
BOND 54 30 33 1 C14 N5
BOND 55 30 41 1 C14 C23
BOND 56 32 33 1 C15 N5
BOND 57 32 34 7 C15 C16
BOND 58 32 40 8 C15 C22
BOND 59 33 65 1 N5 H23
BOND 60 34 35 8 C16 C17
BOND 61 34 66 1 C16 H24
BOND 62 35 36 1 C17 C18
BOND 63 35 37 7 C17 C19
BOND 64 36 67 1 C18 H25
BOND 65 36 68 1 C18 H26
BOND 66 36 69 1 C18 H27
BOND 67 37 38 1 C19 C20
BOND 68 37 39 8 C19 C21
BOND 69 38 70 1 C20 H28
BOND 70 38 71 1 C20 H29
BOND 71 38 72 1 C20 H30
BOND 72 39 40 7 C21 C22
BOND 73 39 73 1 C21 H31
BOND 74 40 42 1 C22 N6
BOND 75 41 42 1 C23 N6
(base)
I also encountered this problem “Atom type of DU does not shown up in PARMCHK.DAT”, and understood the reason and solution based on your conversation, thank you very much
Hello!
Thank you so much for this tutorial! I have a similar situation as above here where I had to proceed with gaff atom types instead of amber atom types due to the chemistry of one of the atoms in my ligand (sp2 imine nitrogen).
I am having an issue generating the appropriate frcmod files to load ff14SB parameters from input files written with gaff atom types. In the tutorial, I noticed that the gaff frcmod files you generated from your amber input files contain the lines (same as c3-c -o , penalty score= 0.0). Correct me if im wrong, but this is essentially telling tleap to match the amber parameters with the gaff parameters.
In my case, I do not see these remarks in the frcmod files i generated for my gaff ligand and wonder how did you work your way around this. Below are the error files from tleap I get when trying to generate topology files for the custom amino acid flanked by two residues.
P.S. I am using AMBER22
Thanks for the help and support!
Javier Sanlley
tleap error:
Error: Could not find bond parameter for atom types: C – n
> for atom C at position -14.036000, 22.450000, -31.516000
> and atom N at position -14.822000, 21.367000, -31.604000.
>
> Error: Could not find bond parameter for atom types: c – N
> for atom C at position -13.727000, 20.025000, -33.379000
> and atom N at position -12.634000, 19.262000, -33.506000.
> Checking for angle parameters.
>
> Error: Could not find angle parameter for atom types: O – C – n
> for atom O at position -12.834000, 22.442000, -31.769000,
> atom C at position -14.036000, 22.450000, -31.516000,
> and atom N at position -14.822000, 21.367000, -31.604000.
>
> Error: Could not find angle parameter for atom types: C – n – hn
> for atom C at position -14.036000, 22.450000, -31.516000,
> atom N at position -14.822000, 21.367000, -31.604000,
> and atom H at position -15.804000, 21.448000, -31.377000.
>
> Error: Could not find angle parameter for atom types: C – n – c3
> for atom C at position -14.036000, 22.450000, -31.516000,
> atom N at position -14.822000, 21.367000, -31.604000,
> and atom CA at position -14.366000, 20.032000, -31.985000.
>
> Error: Could not find angle parameter for atom types: CX – C – n
> for atom CA at position -14.678000, 23.764000, -31.044000,
> atom C at position -14.036000, 22.450000, -31.516000,
> and atom N at position -14.822000, 21.367000, -31.604000.
>
> Error: Could not find angle parameter for atom types: o – c – N
> for atom O at position -14.193000, 20.727000, -34.274000,
> atom C at position -13.727000, 20.025000, -33.379000,
> and atom N at position -12.634000, 19.262000, -33.506000.
>
> Error: Could not find angle parameter for atom types: c – N – H
> for atom C at position -13.727000, 20.025000, -33.379000,
> atom N at position -12.634000, 19.262000, -33.506000,
> and atom H at position -12.326000, 18.698000, -32.725000.
>
> Error: Could not find angle parameter for atom types: c – N – CX
> for atom C at position -13.727000, 20.025000, -33.379000,
> atom N at position -12.634000, 19.262000, -33.506000,
> and atom CA at position -11.821000, 19.222000, -34.715000.
>
> Error: Could not find angle parameter for atom types: c3 – c – N
> for atom CA at position -14.366000, 20.032000, -31.985000,
> atom C at position -13.727000, 20.025000, -33.379000,
> and atom N at position -12.634000, 19.262000, -33.506000.
Hi Javier,
I’m glad you found the tutorial helpful. I believe this is not telling TLEAP to match the Amber parameters with GAFF parameters. As you can see, for that specific angle, values have been assigned to both the force constant and equilibrium angle. It looks like you have a mix between gaff and ff14SB atom types at some stage and because of that tleap is not able to generate the prmtop file. Are you including both frcmod files in the leap script? the one fro ff14SB and tho other for gaff.
Could you share your .ac file with me. I’ll do my best to reproduce the issue and assist you further.
Best wishes,
Carlos
Hi, very helpful guide!
I capped my modified residue, and added the capped atoms to the OMIT_NAME to my mainchain file. During tleap the omitted/capped residues are being added again and I can’t bond my residue to the backbone residues. Do you know how I can prevent the residues from being added?
Dear Dillon,
I’m glad you found the tutorial useful.
What happens if you create a prmtop and inpcrd files for your residue only?
This can be done in the following way:
loadamberprep LTL.prepin
loadamberparams LTL_gaff.frcmod
loadamberparams LTL_ff14SB.frcmod
aa = sequence {LTL}
saveAmberParm aa LTLT.prmtop LTL.rst7
If you see the capped atoms here, something may be missing in the .mc file
If you don’t see the capped atoms here, you may have extra atoms in the PDB file you are using to build your system.
Please let me know if this solves your problem.
Hi Carlos, thank you for this helpful and insightful tutorial!
As I was going through I got a question. What would it happen if we use a higher level of theory to calculate the partial charges than the the one that was used to derive the partial charges of the forcefield?
Would it be safe to say that the ESP of the molecules would not be properly described since the partial charges of that force field are not compatible with that level of theory?
Thank you!
Hi Gerardo,
Thanks for your message. According to the Amber recommendations, using a higher level of theory to calculate partial charges than that used to derive the force field partial charges could lead to discrepancies in the description of the electrostatic potential (ESP) of molecules. This is because the electrostatic potential is sensitive to the distribution of partial charges on atoms.
If the partial charges derived from a higher level of theory differ significantly from those of the force field, the ESP may not be correctly described. This could potentially lead to inaccurate predictions of properties that depend on electrostatic interactions, such as molecular conformations, binding energies or solvation energies.
My recommendation is that it is advisable to follow the methodology outlined in the documentation to ensure consistency and reliability in simulations. If there are compelling reasons to explore a higher level of theory for partial charge calculations, a thorough validation and recalibration of the force field parameters would be necessary to maintain accuracy and compatibility.
Dear Carlos,
I have downloaded the 8CIX structure from the PDB and I want to run molecular dynamics (MD) simulations using AMBER. The peptide in the 8CIX PDB contains four non-standard amino acids: MP8, MVA, MLU, and NZC. I have successfully adjusted the parameters for MP8, MVA, and MLU.
However, I am facing issues with the NZC amino acid. NZC forms peptide bonds with neighboring residues and additionally forms a bond with a glycine residue via its oxygen atom. Specifically, NZC makes two peptide bonds and an additional bond with the carbonyl carbon (C=O) of the glycine backbone.
I am having trouble obtaining the parameters for this additional bond between NZC and glycine. Could you please advise on how to introduce this bond into the AMBER24 program?
Thank you for your assistance.
Best regards,
Hi, I’m glad you managed parameterize the other three residues. For the case of NZC I imagine the problem is related with the fact that this is bound to three residues instead of two as the example in the tutorial. So in these cases what I should do is the geometry optimisation of the structure of the NZC bound to the MP8, both residues with their corresponding capping groups. Perform the calculation of the electrostatic potential charges and the RESP calculation, since at this point you have already obtained the charges for MP8, you can include them in the respq.in file in the same way as the charges of the ACE and the NME. Follow the steps VI and VII of the tutorial but in this last step modify the .mc file to delete not only the atoms of the ACE and NME cap groups but the MP8 as well.
Now you have the .prepin file for your residue, the .frcmod should be generated with the .ac file obtained in the step VI.
Now, the bond between the NZC nitrogen atom and the MP8 Carbon is created during the preparation step using tleap. use the command bond SYSTEM.4.N SYSTEM.3.C. Here, SYSTEM is the name you assign to your system in leap, the 4 and 3 are the number of your residues in the PDB file and N and C are the names of the atoms to be bonded. it’s the same procedure used to create a disulfide bond.
Check my reply to Zhara on “January 11, 2024 at 10:42 am” this can also helps you.
Hope it helps!
Dear Carlos,
Thank you for your prompt response.
The issue we’re encountering is not between NZC and MP8 but between NZC and GLY. The bond between NZC and GLY is an ester bond rather than a peptide bond. Specifically, NZC has two peptide bonds with MP8 and L, and one ester bond with GLY. In the PDB file, the ester bond is represented with the oxygen atom from NZC binding to the carbon atom of the C=O group in GLY.
When I omit the atoms of GLY in the MC file, the prepin file results in multiple ‘X’ signs and lacks all the necessary residues due to identical atom names. Additionally, when I run TLEaP, it adds an OXT atom to GLY. This leads to the modified PDB file showing the same bond twice, appearing aligned but not as a double bond. It also gives me a “one atom does not have a type” error.
Could you please advise on how to properly define that the oxygen atom of NZC binds to the C=O group of GLY? Alternatively, could you please advise on how to run MD simulations for depsipeptides? Is there any example or website you can suggest?
Best
Oh I see, in this case the approach will remain the same as previously mentioned. However, since this is not a regular GLY, you won’t be able to bond it normally to your NZC residue. You’ll need to parameterize it as a new residue, let’s call it GLZ. This residue should be bond to your NZC later you will cutting it using the .mc file and you’ll need to create the bond between it and the NZC in tleap later. The bond with the preceding MLU residue should be created automatically as you include the N and CA atoms in the main chain description in the .mc file.
Your “GLZ” will have a head but no tail, and your NZC and MLU will have both a head and a tail.
Hope this helps, please let me know.
Thanks for this great tutorial! Very useful!!
I got an issue at the step “resp -O -i resp.in -o resp.out -p resp.pch -t resp.chg -q resp.qin -e esp.dat”. The error message looks like “forrtl: severe (64): input conversion error, unit 5, file /home/pz19/Work/Morgan/test/PPP_02/resp.in”
I have deleted the comment lines in resp.in and I am using Amber24. Any insight? Thanks!
Hi Peng,
I’m glad you find this useful.
I understand that working with this file can be a bit tricky. To help, try generating a template for this file using the following antechamber command:
antechamber -fi gout -i LTL_hf.log -bk LTL -fo ac -o LTL.ac -c rc -c resp -at amber
This command is similar to the one you’ll use later to generate the .ac file. However, the key here is to obtain the ANTECHAMBER_RESP1.IN outfile. Rename this file to resp.in, make the necessary modifications, and continue with the fitting charges procedure as outlined in the tutorial, “Restrained Fitting of the Partial Charges to the Electrostatic Potentials (RESP Calculation)”. Compare your file with the generated file and you will see the error you had.
Use the modified resp.in file later with the following command:
resp -O -i resp.in -o resp.out -p resp.pch -t resp.chg -q resp.qin -e esp.dat
Best wishes!
Thank you so much!
Hi Carlos
Following your suggestions, I was able to proceed successfully. Thanks again!
I have one additional question regarding the ” MAIN_CHAIN C3″ in LTL.mc. I am not sure I understand this. Does this mean that C3 connect the head N1 and tail C8? In a general case, should I look for the shorted path that connects the head and tail? Thanks!
Best wishes
Peng
Hi Peng,
Yes, C3, the CA atom, is linked to the N1 head and C8 tail atoms. In general, these three atoms form the backbone of the amino acid.
N1-C3-C8
N-CA-C
HEAD-AlphaCarbon-TAIL
Best wishes,
Carlos
“Conveniently the error in the HF/6-31G* calculation is close to the difference between the charge distribution in gas phase and that in solution “. This link is not working.
Hi Oceanwaves,
It should now work. Thanks for reporting this.
Best wishes,
Carlos
Hey Carlos,
Thank for the amazing tutorial. Been seeking for something like this because these of Amber are quite old !
I have followed a slightly modified path, namely
antechamber -i LIG.pdb -fi pdb -o LIG.gjf -fo gcrt -gv 1 -ge LIG.gesp -nc -2 -pf y
run Gaussian09 on input
antechamber -i LIG.gesp -fi gesp -bk LIG -fo ac -o LIG.ac -c resp -eq 2 -at gaff2 -nc 2 -pf y -rn LIG
prepgen -i LIG.ac -o LIG.prepc -rn LIG
parmchk2 -i LIG.ac -f ac -o LIG.frcmod -a Y -p $AMBERHOME/dat/leap/parm/parm10.dat
Mind sharing your views on it? Particularly in terms of running antechamber on .gesp and picking -c resp rather than what you’ve shown.
Thanks, cheers
Dear Mareczeq,
Thank you for your message. The charge deviation process depends on the specific use case for your simulations. For instance, if you’re planning to run long simulations or performing Free Energy Perturbation calculations, I recommend following the Amber recommendation. This approach provides a more accurate charge deviation model.
On the other hand, if you’re using an approximate method like MM/GBSA with fewer accuracy requirements, a less complex charge deviation model might be sufficient, especially when parameterizing multiple ligands.
Please keep in mind that this is just my opinion, and the best approach depends on your specific application.
Best wishes,
Carlos
I am currently utilizing your tutorial for one of my projects, which involves a protein that contains the SGB residue. I am specifically focusing on the SGB residue as outlined in your tutorial. I have successfully generated the esp.dat file; however, I am encountering difficulties at this stage. The SGB residue features one NH2 group and a C=O group, but it lacks the ACE and NME sections. I am uncertain about how to proceed in obtaining the partial charges and advancing to the next steps. I would greatly appreciate your assistance.
Hi Kandasamy
Could you please provide more context? The ACE and NME groups are added to link your residue to the previous and next residues in the sequence. Are you referring to this residue? https://www.ebi.ac.uk/pdbe/entry/pdb/5fpq/modified/SGB
If that is the case I think what you need to do is to attach the ACE group after the nitrogen and replace the OH group by the NME group.
Best wishes,
Carlos
Thank you so much, yes, it is SGB residue, can you do the tutorial of this pdb.
I have adhered to your instructions; however, I am still unable to resolve the issue. I cannot pinpoint the exact mistake, as I encounter different errors each time. The process functions correctly up to step VII (prepgen), and step VII.2 proceeds without any problems. Nevertheless, I am experiencing an error at step VIII. Despite your guidance to maintain consistent naming in both the prepin and pdb files, I did but the issue persists. In the command, I am not able to attached my files, Can you help me to solve this issue
Hi Kandasamy,
Exactly which error are you obtaining in the step VIII. Are you trying to build the system only using the keyword sequence, as in the tutorial, or are you loading a pdb structure which contains the SGB residue?
Please try to explain me your error and procedure as detailed as possible. Only in this way I’ll be able to help you.
Best wishes,
Carlos
Amazing tutorial! Very few tutorials address parameterizing residues that are a part of a chain and how to apply capping groups. Very helpful!
I do have a question: Utilizing the capping groups, I am able to have my custom residue form bonds with adjacent standard residues. Bonds within the custom residue are now recognized, but any bonds, dihedrals, etc that form across the head/tail atom appear to be missing parameters? (Bonds/angles that contain atoms from both the custom and standard residues)
ie:
Could not find bond parameter for atom types: Os – c3
Could not find angle parameter for atom types: Os – c3 – h2
** No torsion terms for atom types: c3-os-Cg-H2
For further information, these original residue uses the glycam forcefield.
Thanks!
Hi Rokas,
I’m glad you found the tutorial useful. This information used to be in the generated frcmod files. Have you tried generating this file using the GLYCAM_06EPb.dat file instead of gaff for example?
I hope this solves the problem, let me know.
Best wishes,
Carlos
Hello Carlos. I am working on a cyclic peptide ligand. The sequence is YSTCDFIM, where the Met is the C terminal. The CYS side chain forms thiolactone with C-terminal to form a cycle. In this case, how should I custom the residue? Thank you very much for your time and assistance!
Hi Zeyu
This is a modification of a previous reply on a similar query. I hope it works for you:
Thank you for your interest in this tutorial. If you wish to parameterize a residue (MET, MEC from now) bonded to a custom CYS residue (CYC from now), likely through the sulfur, the recommended approach is to begin with an initial structure that includes both residues bonded to each other. Additionally, ensure that each residue has its corresponding cap termini residues ACE and NME.
your initial structure should looks like this:
(ACE and NME capping groups)-CYC—MEC-(ACE capping group)
You will need to create two different parameters files for each residue and create the — bond between CYC and MEC using tleap. I’ll explain this at the end of this message.
If you follow this approach, you can parameterize your custom Cys (CYC) and your custom Met (MEC).
Charges:
To obtain charges for each atom in the RESP program, ensure the preparation of corresponding resp.in and resp.qin files incorporating charge values for atoms in the ACE and NME residues, as demonstrated in the tutorial. You only need one file for the whole (ACE and NME capped)-CYC—MEC-(ACE capped) molecule.
The main distinction arises when creating each unit without additional atoms. For example, to create the CYC residue for Amber recognition you need to specify the exclusion of
all the atoms that don’t belong to the CYC residue, you need to omit all the atoms in MEC and in the three cap termini groups. This is made by indicating their names in the main chain file (CYC.mc file). Your CYC.mc file should resemble the following:
HEAD_NAME N1
TAIL_NAME C8
MAIN_CHAIN C3
OMIT_NAME C1
.
.
.
OMIT_NAME H18
PRE_HEAD_TYPE C
POST_TAIL_TYPE N
CHARGE 0.0 (the charge of your residue)
Adapt it accordingly to your system. List each atom you wish to exclude from your residue by employing the OMIT_NAME keyword, as exemplified in the LTL.mc example file.
Repeat the same procedure for MEC, your MEC.mc file must include all the atoms that don’t belong to the MEC residue. Here you will need to omit all the atoms in CYC and cap termini groups.
Proceed with the rest of the tutorial to generate the frcmod and prepin files.
When preparing your system using leap, remember to bond the two atoms between CYC and MEC with the following line:
bond SYSTEM.XX.AA SYSTEM.YY.BB
Here, SYSTEM is the name you assign to your system/unit on tleap, XX denotes the residue number of CYC and AA (SG, for cysteines) is the name you gave to the sulfur atom bonded to the atom named BB (C, for Mets) in the residue number YY of your “MEC”
I hope this information helps you with the parameterization of your custom residue.
Hi Carlos,
Many thanks for your detailed reply. When I create mc. file for Met residue, is S or N the POST_TAIL_TYPE?
Hi Zeyu,
POST_TAIL_TYPE in your case for the MET residue is S (the carbonyl group is bonded to) and the PRE_HEAD_TYPE C (the amine group is bonded to the ACE capping group)
Best wishes,
Carlos
Hi Carlos,
Thanks for this excellent tutorial. Following it, I was able to parameterize LTL. To test it, I generated a residue xml forcefield file and used it to simulate a protein with a LTL using ISOLDE in ChimeraX (which uses the openmm engine). The simulation ran without error. However, the connections of LTL with the previous and next residues are not correct, although the covalent bonds do exist. The peptide bonds are distorted. For example, the carbonyl oxygen in LTL is almost co-linear with the carbon in LTL and the nitrogen from the previous residue. The peptide bond with the next residue shows similar distortions. For some reason, the capping didn’t seem to ensure the correct bond formation? I am not sure if I did something wrong, because I had to convert the parameters to the xml format.
Thanks,
Xuewu
Hi Xuewu,
I’m glad you found the tutorial useful. When you followed the tutorial, did you get the same result files as in the tutorial? Also, have you tried minimising the LTL residue using Amber? Because if you are able to reproduce the files, and if you do not observe the same co-linearity problem you mentioned, you probably have a problem with the way you generated the residue xml forcefield file.
Please let me know.
Best wishes,
Carlos
Hi Carlos,
You are right, it the xml file that didn’t match the Amber files (atoms names and types were not correctly converted). After correcting this, it worked as expected. Thank you so much again for this great tutorial!
Xuewu
Hi Carlos! First, thank you so much for this amazing tutorial.
I am trying to parametrize some new non-canonical amino acids and while this tutorial has been really helpful for some of them, I am having trouble adapting it to others, especially when it comes to non canonical amino acids (NCAAs) derived from aromatic amino acids.
Right now im working on the parameters for a NCAA derived from phenylalanine. First with tleap I generated the capped original aminoacid { ACE PHE NME }. The problem is when I observed this pdb file with pymol I noticed that NME is actually forming a bond with the aromatic ring. When this pdb file is transformed into Mol2 file, the aromatic ring appears broken (atom that was forming a bond with NME no longer forms bonds within the ring). I tried to fix “by hand” in pymol, just re-generating the hydrogens that were forming the bond so that they would not interact, but it did not help. I also tried generating the mol2 with obabel to see if anything would change but the result was the same.
Do you have any tips on how I could work around this problem?
Thank you!
Hi Rosa,
I’m glad the tutorial has been helpful for you. I generated the amino acid sequence (ACE PHE NME) using tleap. When I visualize it from the prmtop and rst7 files, there is no problem.
Please try the following commands on tleap:
source leaprc.protein.ff14SB
aa = sequence {ACE PHE NME}
saveAmberParm aa aa.prmtop aa.rst7
However, when I convert it into a pdb file using ambpdb (`ambpdb -p aa.prmtop aa.pdb`) and visualize it in PyMol, I see the same issue you mentioned. What you are likely observing is a visualization artifact. In my case, I notice that the HD2 atom from the ring is bonded to the N atom in the NME group, while the H from the amino group is bonded to the CD2 atom in the aromatic ring.
I assume this wouldn’t be an issue if you optimize the geometry following the “II) Geometry Optimization” step. If this continues to be a problem, I recommend trying to rotate the side chain of your NCAA derived from phenylalanine manually. After doing this rotation, the geometry optimization will become easier, and during visualization, the bonds you mentioned should disappear.
I hope it helps!
Hi Carlos,
Thanks for the tutorial. I was trying to replicate the same procedure with NME and ACE capped modified amino acid. In the RESP charge fitting section, I did the initial two steps, now I’m stuck at third step.
how will I get this resp.qin file? Do I need to optimize NME and ACE separately using gaussian and later use espgen to derive charges?
My overall structure has 34 atoms, for which i have resp.in file. Could you help me with this issue?
Thanks
Manisha
Hi Manisha,
If you have the resp.in file, you’ve already completed the hard part! The resp.qin file is simply a file where you will write 0.000000 values for each charge you want to fit from the electrostatic potential you generated in the previous step. If your molecule has 34 atoms, your file will contain 5 lines (4 with 8 values and 1 with only two values). Be sure to replace the 0.000000 values with those in ‘Figure 2. ACE y NME partial charges in the amber forcefield’ as the charge of the rest of the atoms in the molecule will be fitted to conserve the charge provided in the resp.qin file. Compare the resp.qin file provided in the tutorial with the charge values in Figure 2 remember that:
Each value here (resp.qin file) corresponds to an atom in the resp.in file. Thus, if a value in the resp.qin file is different from 0.0000000, a -1 should be written in the second column for that atom in the resp.in file. Comparing the resp.in and the resp.qin files can enhance your understanding.
I hope this helps.
Hi Carlos!
Thanks for the reply. I followed this and was able to generate all the required files. But I’m getting error in the last step while trying to generate topology and coordinate files. It says can’t find bond parameters, angle parameters or torsion terms for various atom types.
How should I resolve this error?
Hi Manisha,
I’m glad you were able to get on with the tutorial. Did you generate the frcmod files with gaff? did you delete the lines with the ATTN messages? Compare the missing parameters with those in the generated frcmod files, this will help you to identify the step where the error occurs.
Best wishes.
Hi CarlosRamosG,
I am following the tutorial for threonine phosphate and at the first I face errors. For amber24 the commands are different?
the first error I got is:
resp -o -i resp.in -o resp.out -p resp.pch -t resp.chg -q resp.qin -e resp.dat
unknown flag: resp.in
usage: resp [-O] -i input -o output -p punch -q qin -t qout -e espot -w qwts -s esout
Hi Fatemeh,
I think the error is related to the fact that the ‘-o’ flag should be in capital letters ‘-O’. If I use the ‘-o’ flag I obtain the same error you reported.
Best wishes.
Hi Carlos,
Thank you for sharing this very useful article.
I feel like something might not be optimal if we do:
> loadamberparams LTL_gaff.frcmod
> loadamberparams LTL_ff14SB.frcmod
because ff14SB parameters are overriding GAFF parameters.
If the conjugated structure on this residue is very different from peptide, then GAFF actually provides better description.
What do you think? Looking forward to hear your opinions.
Ray
Hi Raymond,
Thanks for your comment, you are right. If you have to use a mixed approach, try to combine both files in a way that describes the system in a way that best suits your needs.
Best wishes.
Hi Carlos,
Thank you for sharing this! I’m trying to model a modified residue attched to a natural residue (LYS) using the FF14SB force field, but when I run tleap, I get the following error:
“Could not find angle parameter for atom types: CT – C – CX.”
I’m now trying to find an appropriate angle parameter for these atoms, but I couldn’t locate it in either parm10.dat or GAFF. I came across a paper suggesting that if the angle parameter for A–B–C is missing, one possible approach is to estimate it by averaging the parameters for A–B–A and C–B–C. However, in my case, I couldn’t find the angle parameter for CX–C–CX, although CT–C–CT is available.
What would be the best strategy in this case? I would appreciate any suggestions.
Hi Jeff,
Could you provide more information about the procedure you used to create your custom residue? Is this happening only when you try to bond it to LYS, or the problem also exists if you try to bind it to any other residue? The frcmod files, containing the angle parameters, need to be obtained with the capped structure. If you followed this tutorial the problem could be related to a bad main chain file definition, the .mc file. Amber will only recognize and and bond ‘automatically’ your residue to the previous and next residues in the sequence if you removed properly all the atoms in the cap termini groups during the prepin file generation. Make sure you included all of them using the OMIT_NAME keyword and also check your HEAD_NAME, TAIL_NAME, MAIN_CHAIN, PRE_HEAD_TYPE and POST_TAIL_TYPE are properly assigned in the .mc file. This is pretty much every thing I can say now taking into account the information you provided before.
I hope it helps you to find a solution, best wishes.
Hi Carlos,
Thanks for your reply. I followed the tutorial, but the only difference is how I assigned charges to the atoms—I used Antechamber for this.
I’m working with a non-natural residue that needs to be attached to LYS. In this case, should I cap both ends of the residue, or is capping one end enough?
Also, how can I keep the hydrogen attached to the non-bonding end of my residue? When I cap the non-bonding end, I have to remove the hydrogen to add the NME cap, but I’m not sure how to add the hydrogen back after removing the cap. Maybe this is where the problem lies.
Here is my mc file:
HEAD_NAME C11
TAIL_NAME S
MAIN_CHAIN C10
MAIN_CHAIN C9
MAIN_CHAIN C8
MAIN_CHAIN C7
MAIN_CHAIN C6
MAIN_CHAIN C5
MAIN_CHAIN C4
MAIN_CHAIN C3
MAIN_CHAIN C2
OMIT_NAME C
OMIT_NAME C1
OMIT_NAME O
OMIT_NAME H
OMIT_NAME H1
OMIT_NAME H2
OMIT_NAME N
OMIT_NAME C12
OMIT_NAME H23
OMIT_NAME H24
OMIT_NAME H25
OMIT_NAME H26
PRE_HEAD_TYPE C
CHARGE 0.0
and this is the ac file for my residue:
CHARGE 0.00 ( 0 )
Formula: H27 C13 N1 O1 S1
ATOM 1 C UNL 1 -1.073 -10.383 -0.947 -0.142100 CT
ATOM 2 C1 UNL 1 -1.574 -8.983 -0.794 0.432700 CT
ATOM 3 H UNL 1 -0.512 -10.676 -0.034 0.073700 HC
ATOM 4 H1 UNL 1 -1.917 -11.090 -1.092 0.073700 HC
ATOM 5 H2 UNL 1 -0.397 -10.442 -1.825 0.073700 HC
ATOM 6 S UNL 1 -0.379 -7.653 -0.539 -0.268200 S
ATOM 7 C2 UNL 1 -1.560 -6.260 -0.592 -0.021300 CT
ATOM 8 O UNL 1 -2.774 -8.759 -0.836 -0.436600 OS
ATOM 9 C3 UNL 1 -0.798 -4.950 -0.401 -0.072400 CT
ATOM 10 H3 UNL 1 -2.069 -6.240 -1.580 0.084700 H1
ATOM 11 H4 UNL 1 -2.296 -6.369 0.234 0.084700 H1
ATOM 12 C4 UNL 1 -1.746 -3.746 -0.418 -0.079400 CT
ATOM 13 H5 UNL 1 -0.263 -4.980 0.575 0.052200 HC
ATOM 14 H6 UNL 1 -0.048 -4.842 -1.216 0.052200 HC
ATOM 15 C5 UNL 1 -0.966 -2.441 -0.221 -0.078400 CT
ATOM 16 H7 UNL 1 -2.283 -3.711 -1.392 0.042700 HC
ATOM 17 H8 UNL 1 -2.493 -3.854 0.399 0.042700 HC
ATOM 18 C6 UNL 1 -1.901 -1.227 -0.224 -0.079400 CT
ATOM 19 H9 UNL 1 -0.425 -2.482 0.750 0.041200 HC
ATOM 20 H10 UNL 1 -0.221 -2.333 -1.041 0.041200 HC
ATOM 21 C7 UNL 1 -1.104 0.067 -0.027 -0.079400 CT
ATOM 22 H11 UNL 1 -2.444 -1.182 -1.194 0.040700 HC
ATOM 23 H12 UNL 1 -2.642 -1.332 0.598 0.040700 HC
ATOM 24 C8 UNL 1 -2.026 1.291 -0.023 -0.079400 CT
ATOM 25 H13 UNL 1 -0.557 0.018 0.940 0.040200 HC
ATOM 26 H14 UNL 1 -0.363 0.170 -0.852 0.040200 HC
ATOM 27 C9 UNL 1 -1.216 2.579 0.169 -0.078400 CT
ATOM 28 H15 UNL 1 -2.575 1.342 -0.989 0.040700 HC
ATOM 29 H16 UNL 1 -2.763 1.193 0.804 0.040700 HC
ATOM 30 C10 UNL 1 -2.130 3.810 0.175 -0.083400 CT
ATOM 31 H17 UNL 1 -0.665 2.527 1.133 0.040700 HC
ATOM 32 H18 UNL 1 -0.479 2.675 -0.660 0.040700 HC
ATOM 33 C11 UNL 1 -1.317 5.094 0.362 0.161800 CT
ATOM 34 H19 UNL 1 -2.683 3.862 -0.788 0.046700 HC
ATOM 35 H20 UNL 1 -2.865 3.720 1.006 0.046700 HC
ATOM 36 N UNL 1 -2.211 6.257 0.377 -0.826200 N
ATOM 37 H21 UNL 1 -0.767 5.033 1.327 0.028200 H1
ATOM 38 H22 UNL 1 -0.581 5.189 -0.469 0.028200 H1
ATOM 39 C12 UNL 1 -1.451 7.488 0.594 0.153100 CT
ATOM 40 H23 UNL 1 -2.693 6.331 -0.550 0.355800 H
ATOM 41 H24 UNL 1 -0.933 7.455 1.577 0.028700 H1
ATOM 42 H25 UNL 1 -0.705 7.640 -0.217 0.028700 H1
ATOM 43 H26 UNL 1 -2.146 8.354 0.601 0.028700 H1
BOND 1 1 2 1 C C1
BOND 2 1 3 1 C H
BOND 3 1 4 1 C H1
BOND 4 1 5 1 C H2
BOND 5 2 6 1 C1 S
BOND 6 2 8 1 C1 O
BOND 7 2 8 1 C1 O
BOND 8 6 7 1 S C2
BOND 9 7 9 1 C2 C3
BOND 10 7 10 1 C2 H3
BOND 11 7 11 1 C2 H4
BOND 12 9 12 1 C3 C4
BOND 13 9 13 1 C3 H5
BOND 14 9 14 1 C3 H6
BOND 15 12 15 1 C4 C5
BOND 16 12 16 1 C4 H7
BOND 17 12 17 1 C4 H8
BOND 18 15 18 1 C5 C6
BOND 19 15 19 1 C5 H9
BOND 20 15 20 1 C5 H10
BOND 21 18 21 1 C6 C7
BOND 22 18 22 1 C6 H11
BOND 23 18 23 1 C6 H12
BOND 24 21 24 1 C7 C8
BOND 25 21 25 1 C7 H13
BOND 26 21 26 1 C7 H14
BOND 27 24 27 1 C8 C9
BOND 28 24 28 1 C8 H15
BOND 29 24 29 1 C8 H16
BOND 30 27 30 1 C9 C10
BOND 31 27 31 1 C9 H17
BOND 32 27 32 1 C9 H18
BOND 33 30 33 1 C10 C11
BOND 34 30 34 1 C10 H19
BOND 35 30 35 1 C10 H20
BOND 36 33 36 1 C11 N
BOND 37 33 37 1 C11 H21
BOND 38 33 38 1 C11 H22
BOND 39 36 39 1 N C12
BOND 40 36 40 1 N H23
BOND 41 39 41 1 C12 H24
BOND 42 39 42 1 C12 H25
BOND 43 39 43 1 C12 H26
Hi Jeff,
I have checked your structure. One problem is that it doesn’t contain any peptide bonds. This means that when you try to remove the NME capping group, the generated molecule won’t be able to bond to your LYS. The capping groups mimic the amino acid that you are trying to attach to this fragment. If you want to bond UNL to a lysine residue through Lysine’s C-terminal, you will need to add an ACE capping group to the UNL residue and attach it to the UNL N atom, and likely your MAIN_CHAIN atom is C10. i think your structure should look something like (UNL_including_N)-(ACE)
If this is not the case, I have some additional questions.
Does UNL need to be attached to Lys by the N- or C-terminal? Is UNL at the end of a chain? If so, would you like it to be simulated as capped or charged?
If you only need to bond this residue through one of its sides, cap that side. Those are the atoms from the capping group that you need to delete later using the OMIT_NAME keyword in the .mc file.
I didn’t understand this part:
“Also, how can I keep the hydrogen attached to the non-bonding end of my residue? When I cap the non-bonding end, I have to remove the hydrogen to add the NME cap, but I’m not sure how to add the hydrogen back after removing the cap. Maybe this is where the problem lies.”
If you are not planning to bond anything to the C or N end of your structure, you simply need to model it as COO- or NH₃⁺, respectively.
Best wishes.
Hi Carlos,
Thank you for the explanation. UNL is not charged. Actually my structure should look like (LYS – UNL_without N). I want to connect the C-terminal of LYS to the C-terminal of UNL (specifically the C11 atom).
I thought I needed to cap the bonding side of UNL with NME because you mentioned in the tutorial that the N atom should be the head atom. Since in tleap, I use the sequence sequence {LYS UNL}, the head side of UNL becomes the bonding side in the sequence. So, I assumed I should use NME on the bonding side.
But I think I may have misunderstood. Are you saying that the cap group on the bonding side should actually be ACE? In other words, the type of cap group depends on the type of atom in the next residue, correct? I’m asking because I plan to simulate additional residues in place of LYS in the future, and I want to ensure I use the correct approach in those cases.
Regarding the part I mentioned earlier:
“Also, how can I keep the hydrogen attached to the non-bonding end of my residue? When I cap the non-bonding end, I have to remove the hydrogen to add the NME cap, but I’m not sure how to add the hydrogen back after removing the cap. Maybe this is where the problem lies.”
I was trying to say that I have a hydrogen attached to the sulfur atom in UNL (on the non-bonding side). When I cap that side with ACE, I need to attach ACE to the sulfur, which means I have to remove the hydrogen. After removing the ACE cap, I’m not sure how to add the hydrogen back to the sulfur atom, since that end is not supposed to connect to any other residue.
However, I think you were saying that I don’t need to cap the non-bonding side at all, right? In that case, I wouldn’t need to remove the hydrogen, so this issue would be resolved.
Thanks!
Hi Jeff,
First things first, technically speaking, you are not creating a custom residue. This is one of the reasons why this tutorial requires a workaround. It’s simply something bonded to a LYS; there is no peptide backbone between the UNL and the LYS.
The structure that you use to parameterise UNL should be as similar as possible to the final structure that you want to simulate. For example, if a carbonyl group is going to be bonded to the C11 atom, you need to include an ACE group bonded to C11 in the starting structure.
Exactly, if you have a SH group at the end of the chain then parameterize it like that, there is no reason to include an ACE group bonded to the sulfur.
Hi Carlos,
I just tried building my sequence by capping only the ACE group at the bonding side of my residue (UNL), but I still encountered the same error. It appears that the ff14SB and GAFF force fields do not contain the angle parameter for the carbon atoms I mentioned (CX-C-CT). I believe we might need to approximate some parameters by identifying similar ones in such cases. I’ve been looking for a solution, but so far, I have not found one.
Best.
Hi Jeff,
I think the solution is much simpler: try generating the frcmod for UNL using a structure containing UNL bonded to a regular amino acid. An ALA should suffice (UNL-C11-ALA). I’m pretty sure the parameters for that angle are included in the GAFF parameters.
Best wishes,
Hi Carlos,
Thanks for sharing this! I’m trying to model a modified residue using the ff14SB force field, but when I run tleap, I get the following error:
“Could not find angle parameter for atom types: CT – C – CX.”
I’m now trying to find an appropriate angle parameter for these atoms, but I couldn’t locate it in either parm10.dat or GAFF. I came across a paper suggesting that if the angle parameter for A–B–C is missing, one possible approach is to estimate it by averaging the parameters for A–B–A and C–B–C. However, in my case, I couldn’t find the angle parameter for CX–C–CX, although CT–C–CT is available.
What would be the best strategy in this case? I would appreciate any suggestions or guidance.
Thank you!
Hi Jeff, please take a look to my previous reply.
Hi Carlos,
I am working on a modified residue. I have capped it. I want to know when exactly do we need to decap the residue and how are we supposed to do that?
Hi Saloni,
When you create the main chain file (.mc), you will remove the capping groups. All atoms forming the capping groups must be included in the OMIT_NAME section of it. This will ensure that the generated residue bonds to its adjacent residues during LEAP preparation, as this information will be included in the generated .prepin file.
I hope it helps,
Carlos
Hi Carlos,
Where did you generate the partial charges that were written on each atom in figure 2 from?
I know you placed them in the resp.qin file, but I want to know if they were from the esp output file, and how can I locate the section?
Hi Toyin,
Those are the values from the ff14SB, take a look to the figure 1.2 in this amber tutorial https://ambermd.org/tutorials/advanced/tutorial1/section1.php
A fast and easy way to check the charges from any atom in this forcefield is generating a set of prmtop and rst7 files of the residue/s you are interested in using the following commands on tleap:
source leaprc.protein.ff14SB
aa = sequence {ACE NME}
saveAmberParm aa aa.prmtop aa.rst7
If you load this structure in VMD, you can see the charge of each atom by clicking on it after pressing 1. To change the display from the resname:resid:atom name to the charge value, go to the VMD menu, select Graphics > Labels > Atoms Properties, and change the format from %R%d:%a to %q.
In this way you will be able to see the values in figure 2 of this tutorial.
Best wishes,
Carlos
Hi Carlos,
I successfully run your tutorial L-tert-Leucine and at the end parameters for all LT as well as ACE and NME also generated which we omitted here.
Let say I want to generate parameters for an amino acid in Zwitterionic form, Cationic and Anionic form individually, can I use the same method to generate it, If so then how to proceed. Could you please help me on this this matter?
Regards
Abdul Aziz
Hi Abdul,
I’m glad the tutorial was useful to you! If you want to use the amino acid as the C-terminal, carboxylate negatively charged, you can follow the tutorial as you did before, but this time, only include the NME cap terminal group. Later, you will only need to remove the atoms from this group using the main chain file (.mc). That would be the only change from the tutorial. To parameterize it as an N-terminal amino acid with a positively charged amino group, cap it only with the ACE group.
I hope this helps with parameterizing the amino acid in its corresponding C- and N-terminal forms.
Best wishes,
Carlos
Dear Carlos,
Thank you for your kind reply and prompt attention to my issue. Let’s say I want to generate parameters for tryptophanate, that is, there is an NH2 group and a COO- group on that amino acid. Now, if I used this step, as you mention, “If you want to use the amino acid as the C-terminal, carboxylate negatively charged, you can follow the tutorial as you did before, but this time, only include the NME cap terminal group. Later, you will only need to remove the atoms from this group using the main chain file (.mc).” After removing that NME cap, how can I add the O- part to that C=O and get their parameter as I want to make COO- and also vice versa.
Also mention if I want to parametrise a zwitterion, means both NH3+ and COO- then how to do that? It will be very helpful if you help me out on this matter. Thank you in advance.
Regards
Abdul Aziz
Dear Carlos,
Thank you for your kind reply and prompt attention to my issue. Let’s say I want to generate parameters for tryptophanate, that is, there is an NH2 group and a COO- group on that amino acid. Now, if I used this step, as you mention, “If you want to use the amino acid as the C-terminal, carboxylate negatively charged, you can follow the tutorial as you did before, but this time, only include the NME cap terminal group. Later, you will only need to remove the atoms from this group using the main chain file (.mc).” After removing that NME cap, how can I add the O- part to that C=O and get their parameter as I want to make COO- and also vice versa.
Also mention if I want to parametrise a zwitterion, means both NH3+ and COO- then how to do that? It will be very helpful if you help me out on this matter. Thank you in advance.
Regards
Abdul Aziz
Hi Abdul,
I’m not sure if there is a fast way to do that in just one step. But what you can do is to parameterize tryptophanate each positively and degatively charged individually, assigning to each one a unique name, as it’s made for the standard amino acids in the AMBER force field. For example, ALA N terminal is called NALA, while ALA C terminal is called CALA. So, this means you need to follow every step for each one.
Regarding the zwitterionic form, you are facing a small molecule parameterization scenario in this case, so you can follow AMBER’s Sustiva tutorial for parameterization. You don’t need to worry about automatic bonding detection here, so you don’t need to create the prepin file from the main chain file.
Best wishes,
Carlos
See previous comment.
Dear Carlos,
I make a molecule “Let’s say I want to generate parameters for tryptophanate, that is, there is an NH2 group and a COO- group on that amino acid. Now, if I used this step, as you mention, “If you want to use the amino acid as the C-terminal, carboxylate negatively charged, you can follow the tutorial as you did before, but this time, only include the NME cap terminal group. Later, you will only need to remove the atoms from this group using the main chain file (.mc).” After removing that NME cap, how can I add the O- part to that C=O and get their parameter as I want to make COO- and also vice versa.” using this as you said. But the problem comes in sequencing to generate the isolated single tryptophanate parameters that is with NH2-TRP-COO- but what I get when sequence choose CTRP structure means 1 H missing from the NH2 site. Is there anything that I can add H on it later Or what sequence I need to choose ? Can you help me on this matter? Thank you in advance.
Sequence I choose :: trp = sequence { CTRP}
Thanks
Abdul
Hi Abdul,
I’m not sure if I understood your question properly. If you want to simulate free tryptophanate with both ends unprotected—meaning it has a free NH₂ group at the N-terminus and a free COO- group at the C-terminus, as found in isolated amino acids—you don’t need to cap the molecule. You also don’t need to generate the .prepin file because you won’t bind this molecule to any other fragment. This is why the H disappears after you remove the capping group attached to the nitrogen atom. When you use the .mc file to generate the .prepin file, you are creating a fragment that will bond to another fragment through this N atom.
You don’t need to create a sequence to simulate tryptophanate; you need to parameterize it as a small molecule instead of a nonstandard amino acid. Technically, it’s the same, but the parameterization process follows a different logic. My recommendation for that is to follow the AMBER tutorial to parameterize Sustiva (https://ambermd.org/tutorials/basic/tutorial4/create_prmtop.htm). This is what you need for tryptophanate.
I hope this clarifies the issue.
Best wishes,
Carlos
Hi Carlos,
I am not completely certain on how to proceed with the custom residue and the original protein I want to simulate. I have the residue structure of my custom residue (prepin) and missing forcefield parameters (frcmod), how do I combine them with the original protein (or rather replace the original residue) and load them into amber now? Do I just edit the original PDB by hand?
Thanks for the tutorial!
Best wishes,
Blue
Hi Blue,
You need to modify the name of the residue in the PDF file that you are using to build your system in TLEAP. For example, let’s say you want to replace a hydrogen atom with a chlorine atom in a glycine residue.
In your original PDB file, it would look something like this:
ATOM 13 N GLY A 2 4.000 32.907 -13.026 1.00 30.62 N
ATOM 14 CA GLY A 2 2.560 33.214 -13.015 1.00 30.61 C
ATOM 15 C GLY A 2 1.858 32.587 -11.825 1.00 30.01 C
ATOM 16 O GLY A 2 2.429 31.673 -11.175 1.00 31.86 O
ATOM 17 H GLY A 2 4.571 33.413 -12.363 1.00 30.62 H
ATOM 18 HA3 GLY A 2 2.105 32.853 -13.937 1.00 30.61 H
ATOM 19 HA2 GLY A 2 2.420 34.295 -12.990 1.00 30.61 H
Simply edit the column containing the name of your residue in your original PDB file to include the name you gave to create your custom residue (GLC, for example). If the atoms in this new residue have different names, rename them. Here, I’m only changing the HA3 name from GLY to CL in GLC. If your new residue contains more atoms than the one in the PDB file, that won’t be a problem as tleap will add the missing atoms according to the description in your .prepin file.
ATOM 13 N GLC A 2 4.000 32.907 -13.026 1.00 30.62 N
ATOM 14 CA GLC A 2 2.560 33.214 -13.015 1.00 30.61 C
ATOM 15 C GLC A 2 1.858 32.587 -11.825 1.00 30.01 C
ATOM 16 O GLC A 2 2.429 31.673 -11.175 1.00 31.86 O
ATOM 17 H GLC A 2 4.571 33.413 -12.363 1.00 30.62 H
ATOM 18 CL GLC A 2 2.105 32.853 -13.937 1.00 30.61 Cl
ATOM 19 HA2 GLC A 2 2.420 34.295 -12.990 1.00 30.61 H
Let me know if it worked.
Good luck!
Hi Carlos,
Thanks for the quick reply! If I have more atoms than in the original PDB file, do I then have to adjust the atom numbering for the residues following my custom residue as well? And where do I take the coordinates for those extra residues from?
Best wishes,
Blue
Hi Blue,
There’s no need to worry about renumbering, tLEaP will handle that automatically to account for any additional atoms not present in the original PDB structure.
I’m not sure I understood your second question properly, so please correct me if I’m wrong. I’ll assume two things: first, that you already have a protein with a sequence like ACE-ALA-GLY-LYS-MET-NME, for example, and second, that you want to replace the GLY residue with GLC (using the same nomenclature from my previous reply). If that’s the case, you don’t need to manually add coordinates for those extra atoms, though if you meant residues rather than atoms, please clarify so I can give you a more precise answer.
To verify everything is working as expected, try running a tLEaP test to generate the parameter and coordinate files, then visualize them in VMD (or a similar tool). In principle, tLEaP should be able to rebuild the entire side chain of your new residue from the information in your
.prepinfile, much like it automatically adds missing protons based on the amino acid name in the PDB file.Please let me know if it works.
Best
Carlos
Hi Carlos,
Okay, thank you!
For clarification: I parametrized substrate bound to an active site lysine via schiff base as a custom residue so I can paste it into different homologs and simulate the interaction of the protein with the substrate. This means that my custom residue (called it KPS) has more atoms than the original lysine in the pdb (the 3 C’s from the propanal that are now covalently bound). If I paste them in the original pdb, I do not have any existing coordinates for them plus the atom numbering in the pdb changes, so I am unsure about how to do that. Could I “build” another pdb in ChimeraX or a similar program where I insert the custom residue mol2 into the protein instead of the lysine and then take that pdb as an input for tleap?
Thanks a lot for your help!
Best wishes,
Blue
Hi Blue,
Thanks for the clarification. Based on your explanation, there are two options available:
1. Use ChimeraX, as you suggested.
2. Simply rename the LYS residue to KPS directly in the PDB files for the homologs you want to study.
The second option may seem counterintuitive at first, but here’s the reasoning: if you provide an incomplete residue to tleap, it will automatically add the missing atoms based on the information in the force field, your
KPS.prepinfile for this residue (LYSN=C3). Additionally, tleap ignores atom numbering order in the PDB file, so it doesn’t matter if the new residue has more or fewer atoms than the original.To see this in action, try the following test in tleap:
Step 1 – Generate a reference LYS structure:
source leaprc.protein.ff14SBstandard = sequence {ACE LYS NME}
savepdb standard LYS.pdb
Step 2 – Create the system containing KPS:
Copy
LYS.pdbto a new file calledKPS_incomplete.pdb, then replace every instance ofLYSin the residue name column withKPS. If you want to push tleap a little harder, you can delete all the atoms forming the side chain of LYS. tleap will then add these back, along with the covalently bonded propanal.Step 3 – Build KPS in tleap:
loadamberprep KPS.prepinsource leaprc.protein.ff14SB
loadamberparams KPS_ff14SB.frcmod
custom = loadpdb KPS_incomplete.pdb
savepdb custom KPS.pdb
Visualize KPS.pdb
This will show you how tleap handles the renamed residue and fills in any missing atoms automatically.
Just check that tleap is not placing the atoms of the covalent bonded propanal in an unexpected way.
If you need to run multiple homologs in a high-throughput manner, this procedure will help you.
Please let me know if it works.
Best
Carlos